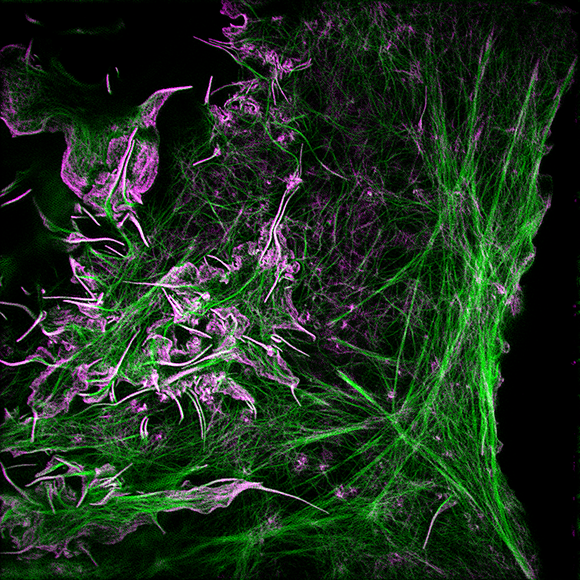
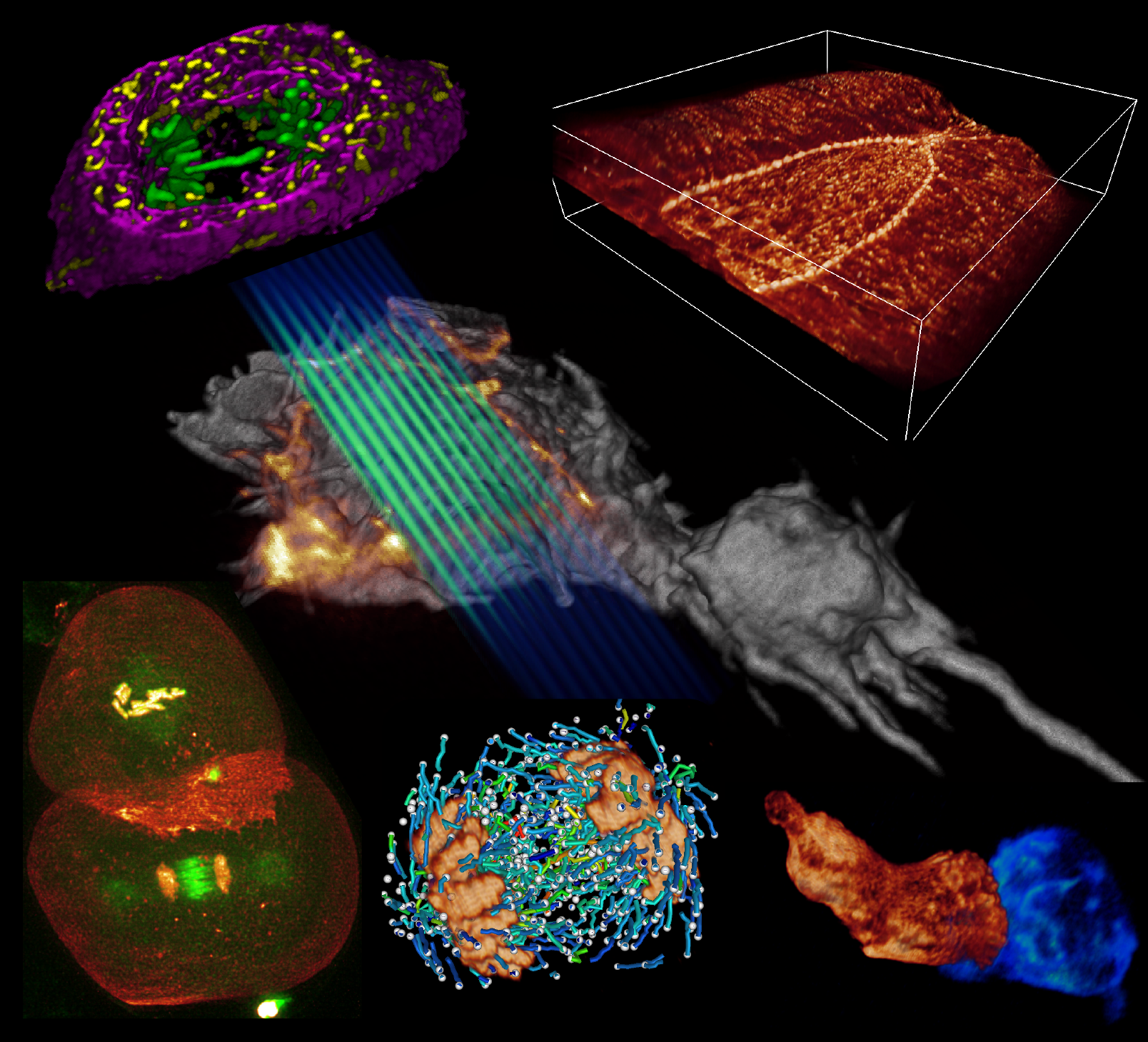
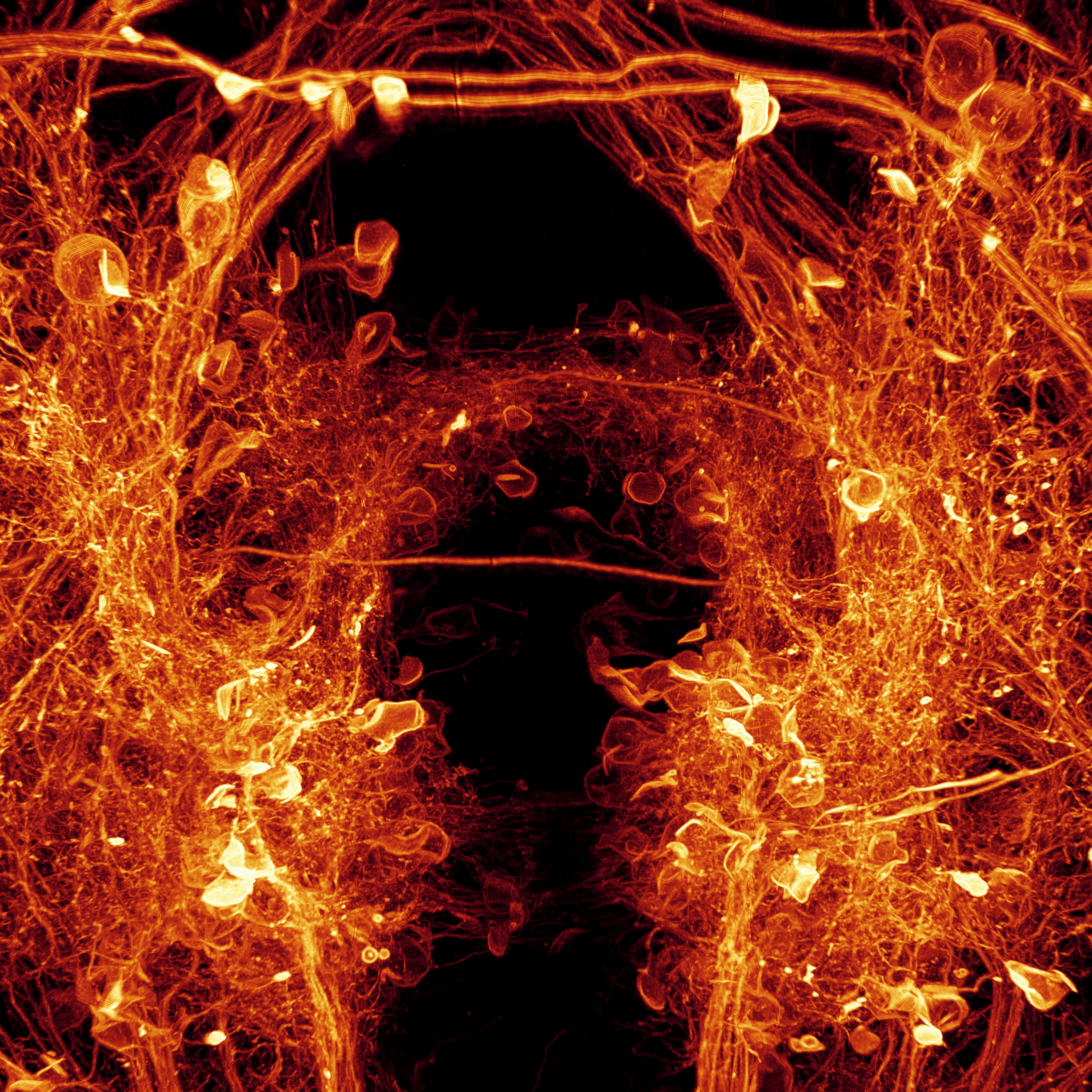
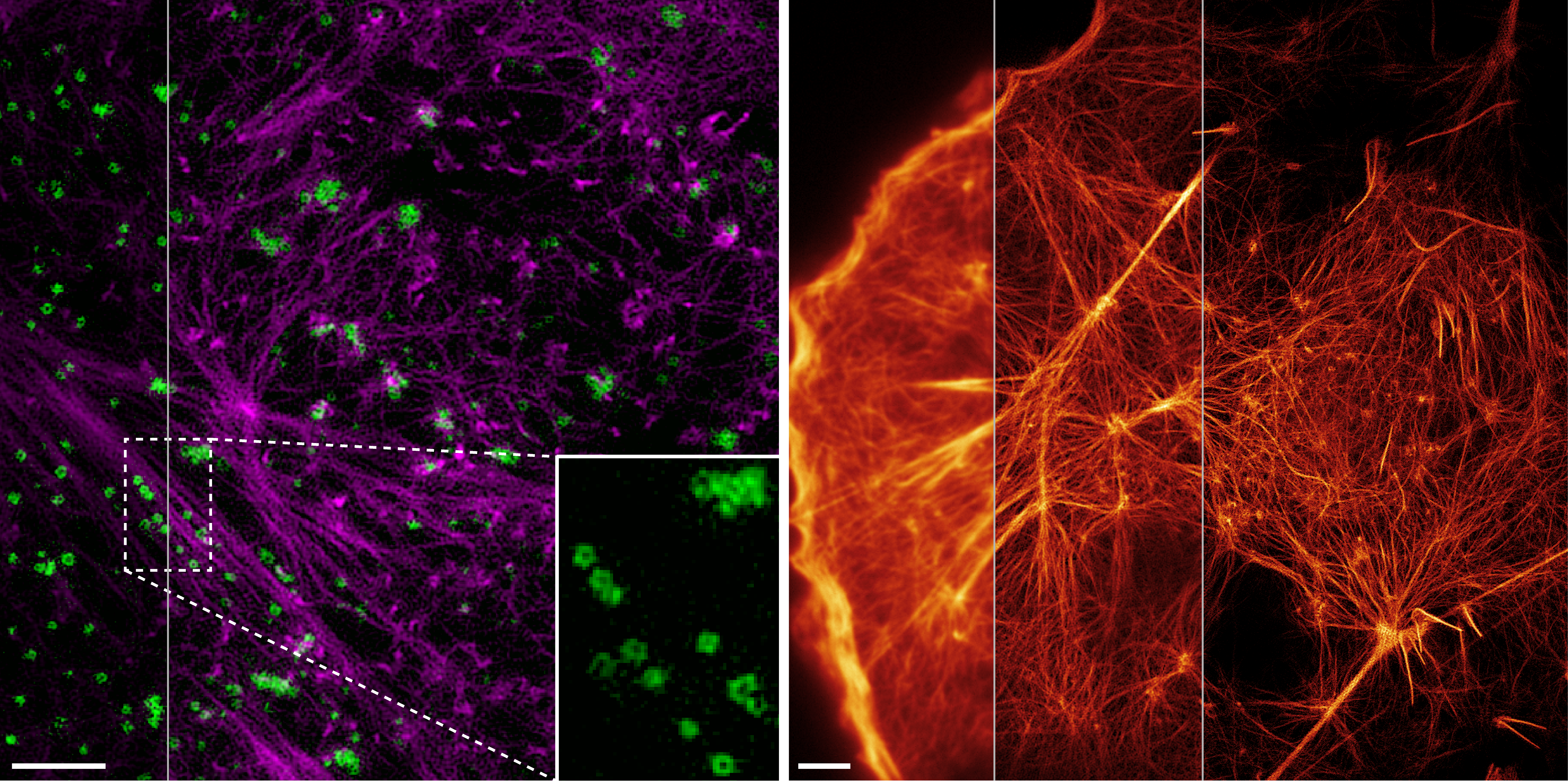
I welcome inquiries from anyone with a Ph.D. in the physical sciences interested in postdoctoral, research specialist, or senior scientist positions involving the development of new bioimaging technologies and their application. Prerequisites are creativity, passion, dedication, competitiveness, and the ability to work collaboratively in a small group. Experience in both optical technology and live cell imaging is desired but not essential. We seek not technology for technology’s sake, but rather those opportunities with the potential to have a broad and deep impact on biomedical research.
Contact me - betzige (at) janelia.hhmi.org
The current group, summer 2015: Tsung-Li, Dong, Wes, Eric, Kai, David
