In the long history of humankind (and animal kind too) those who learned to collaborate and improvise most effectively have prevailed.
Charles Darwin
(1809-1882)
We specialize in developing high-throughput 3D electron microscopy and super-resolution 3D optical microscopy for brain connectomics and cell biology.


I. Super-Resolution Optical Microscopy for Three Dimensions: iPALM & CLEM
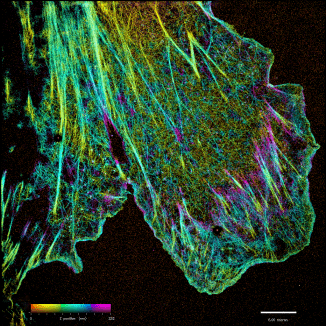
iPALM: Majority of biological problems are 3-dimensional. However, most of the original super-resolution microscopy methods only had high resolution in lateral plane, and suffered substantially poorer resolution in axial (z-) direction. We developed an interferometric photoactivated localization microscopy (iPALM), the combination of photoactivated localization microscopy (PALM) with single-photon, simultaneous multiphase interferometry that provides sub-20-nm 3D protein localization with optimal molecular specificity. Watch a video of the personal story of how PALM was invented by Harald Hess and Eric Betzig.
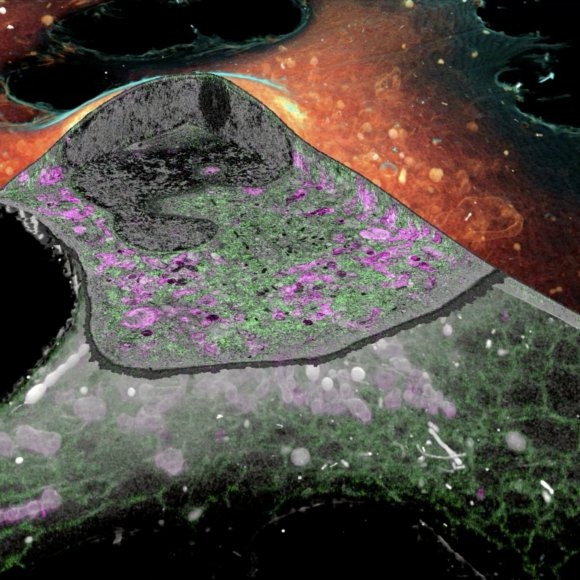
CLEM: Correlative light and electron microscopy (CLEM) is attractive because it exploits two microscopy techniques that give very different and very complementary information. By combining light microscopy (LM) and electron microscopy (EM), one is able to achieve protein specific localization in the context of a global structure. We develop different correlative optical super-resolution and electron microscopy methods optimized for various types of biological problems and sample configurations. With recent advancement in cryo-SR/FIB-SEM technology, the method melds the best of super-resolution fluorescence and electron microscopy to show how protein relate to cell's find structure.
II. High-Throughput Electron Microscopy for Large Volume: FIB-SEM & GCIB-SEM
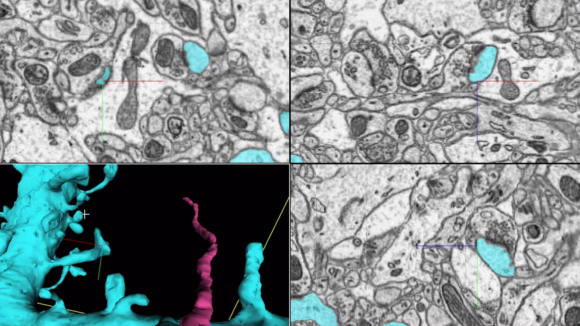
FIB-SEM: Isotropic high-resolution imaging of large volumes provides unprecedented opportunities to advance connectomics and cell biology research. Conventional Focused Ion Beam Scanning Electron Microscopy (FIB-SEM) offers unique benefits such as high resolution (< 10 nm in x, y, and z) and robust image alignment. However, its prevailing deficiencies in imaging speed and duration, cap the maximum possible image volume. Enhanced FIB-SEM 1.0 development started in 2009, with 10 years of effort [Xu et al. 2017; Xu et al. 2020a; Xu et al. 2020b], we have transformed the conventional FIB-SEM from a metrology tool that is unreliable for more than a few days to a robust imaging platform: capable of years of continuous imaging without defects in the final image stack. As a result, the imageable volume of FIB-SEM has been enlarged by more than four orders of magnitude from 103 µm3 to at least 107 µm3 while maintaining an isotropic sampling of 8 x 8 x 8 nm3 voxels. The largest connectome to date has been generated through this enhanced FIB-SEM 1.0 platform [Xu et al. 2020c], where the superior z resolution empowers automated tracing of neuronal processes and reduces the time-consuming human proofreading effort. By trading off imaging speed, the system can readily be operated at even higher resolutions achieving voxel sizes of 4 x 4 x 4 nm3. Moreover, combining with super-resolution fluorescence imaging, CLEM applications at the whole cell level unleash the full potential of intracellular organelle identification with labeling insights. To expand the horizon of this powerful discovery platform, in 2020, Janelia launched a new division, FIB-SEM Technology, led by C. Shan Xu to spearhead the development of enhanced FIB-SEM 2.0 & 3.0.
GCIB-SEM: FIB-SEM relies on single electron beam imaging thus limiting throughput. New multibeam SEMs, like the 61, 91, and soon 331 beam Zeiss MultiSEMs [Riedesel et al. 2019], promise dramatic improvements in acquisition speed with equivalent XY resolution. Unfortunately these multibeam SEMs can only provide such fast imaging for wide area samples which are not compatible with focused ion beam milling. To overcome this limitation, we are developing new ion milling techniques which are compatible with wide area samples. One of these is Gas Cluster Ion Beam (GCIB) milling which uses a broad beam of ionized argon clusters (2000 atoms per cluster, directed at a 30oglancing angle) to mill away the top few nanometers of a tissue’s surface [Hayworth et al. 2018; Hayworth et al. 2019].
To GCIB-SEM image a tissue volume we first cut it into a series of relatively thick sections (100nm – 1µm, also known as “semi-thin” sections) which are collected onto a silicon wafer. The top surface of each section is then imaged (by a single beam or multibeam SEM) and then the entire wafer is subjected to ion milling. This image/mill cycle is repeated as many times as needed to mill through all sections. The result is FIB-SEM-like volume images of each of the semi-thin sections which can then be stitched together with our custom software into a final volume suitable for connectomic tracing.
We are currently working on integrating this wide-area ion milling approach with our recently installed 61-beam Zeiss MultiSEM.
 Overview of GCIB-SEM process.
Overview of GCIB-SEM process.
iPALM Image
In the long history of humankind (and animal kind too) those who learned to collaborate and improvise most effectively have prevailed.
Charles Darwin
(1809-1882)
