Filter
Associated Lab
- Aso Lab (1) Apply Aso Lab filter
- Betzig Lab (115) Apply Betzig Lab filter
- Bock Lab (1) Apply Bock Lab filter
- Clapham Lab (2) Apply Clapham Lab filter
- Fetter Lab (2) Apply Fetter Lab filter
- Harris Lab (7) Apply Harris Lab filter
- Hess Lab (8) Apply Hess Lab filter
- Ji Lab (11) Apply Ji Lab filter
- Lavis Lab (8) Apply Lavis Lab filter
- Lippincott-Schwartz Lab (6) Apply Lippincott-Schwartz Lab filter
- Liu (Zhe) Lab (7) Apply Liu (Zhe) Lab filter
- Magee Lab (2) Apply Magee Lab filter
- Rubin Lab (1) Apply Rubin Lab filter
- Saalfeld Lab (2) Apply Saalfeld Lab filter
- Schreiter Lab (1) Apply Schreiter Lab filter
- Shroff Lab (9) Apply Shroff Lab filter
- Singer Lab (1) Apply Singer Lab filter
- Svoboda Lab (2) Apply Svoboda Lab filter
- Tjian Lab (4) Apply Tjian Lab filter
- Turner Lab (1) Apply Turner Lab filter
Associated Project Team
Publication Date
- 2025 (4) Apply 2025 filter
- 2024 (2) Apply 2024 filter
- 2023 (4) Apply 2023 filter
- 2022 (3) Apply 2022 filter
- 2021 (2) Apply 2021 filter
- 2020 (4) Apply 2020 filter
- 2019 (7) Apply 2019 filter
- 2018 (6) Apply 2018 filter
- 2017 (8) Apply 2017 filter
- 2016 (12) Apply 2016 filter
- 2015 (11) Apply 2015 filter
- 2014 (8) Apply 2014 filter
- 2013 (4) Apply 2013 filter
- 2012 (5) Apply 2012 filter
- 2011 (7) Apply 2011 filter
- 2010 (3) Apply 2010 filter
- 2009 (2) Apply 2009 filter
- 2008 (8) Apply 2008 filter
- 2007 (2) Apply 2007 filter
- 2006 (1) Apply 2006 filter
- 2005 (1) Apply 2005 filter
- 1995 (1) Apply 1995 filter
- 1994 (2) Apply 1994 filter
- 1993 (2) Apply 1993 filter
- 1992 (4) Apply 1992 filter
- 1991 (2) Apply 1991 filter
Type of Publication
115 Publications
Showing 61-70 of 115 resultsThe diffraction limited resolution of two photon and confocal microscope can be recovered using adaptive optics to explore the detailed neuronal network in the brains of zebrafish and mouse in vivo.
The endoplasmic reticulum (ER) is an expansive, membrane-enclosed organelle that plays crucial roles in numerous cellular functions. We used emerging superresolution imaging technologies to clarify the morphology and dynamics of the peripheral ER, which contacts and modulates most other intracellular organelles. Peripheral components of the ER have classically been described as comprising both tubules and flat sheets. We show that this system consists almost exclusively of tubules at varying densities, including structures that we term ER matrices. Conventional optical imaging technologies had led to misidentification of these structures as sheets because of the dense clustering of tubular junctions and a previously uncharacterized rapid form of ER motion. The existence of ER matrices explains previous confounding evidence that had indicated the occurrence of ER “sheet” proliferation after overexpression of tubular junction–forming proteins.
In an interferometer-based fluorescence microscope, a beam splitter is often used to combine two emission wavefronts interferometrically. There are two perpendicular paths along which the interference fringes can propagate and normally only one is used for imaging. However, the other path also contains useful information. Here we introduced a second camera to our interferometer-based three-dimensional structured-illumination microscope (I(5)S) to capture the fringes along the normally unused path, which are out of phase by π relative to the fringes along the other path. Based on this complementary phase relationship and the well-defined phase interrelationships among the I(5)S data components, we can deduce and then computationally eliminate the path length errors within the interferometer loop using the simultaneously recorded fringes along the two imaging paths. This self-correction capability can greatly relax the requirement for eliminating the path length differences before and maintaining that status during each imaging session, which are practically challenging tasks. Experimental data is shown to support the theory.
The inner ear is a fluid-filled closed-epithelial structure whose function requires maintenance of an internal hydrostatic pressure and fluid composition. The endolymphatic sac (ES) is a dead-end epithelial tube connected to the inner ear whose function is unclear. ES defects can cause distended ear tissue, a pathology often seen in hearing and balance disorders. Using live imaging of zebrafish larvae, we reveal that the ES undergoes cycles of slow pressure-driven inflation followed by rapid deflation. Absence of these cycles in mutants leads to distended ear tissue. Using serial-section electron microscopy and adaptive optics lattice light-sheet microscopy, we find a pressure relief valve in the ES comprised of partially separated apical junctions and dynamic overlapping basal lamellae that separate under pressure to release fluid. We propose that this lmx1-dependent pressure relief valve is required to maintain fluid homeostasis in the inner ear and other fluid-filled cavities.
Although fluorescence microscopy provides a crucial window into the physiology of living specimens, many biological processes are too fragile, are too small, or occur too rapidly to see clearly with existing tools. We crafted ultrathin light sheets from two-dimensional optical lattices that allowed us to image three-dimensional (3D) dynamics for hundreds of volumes, often at subsecond intervals, at the diffraction limit and beyond. We applied this to systems spanning four orders of magnitude in space and time, including the diffusion of single transcription factor molecules in stem cell spheroids, the dynamic instability of mitotic microtubules, the immunological synapse, neutrophil motility in a 3D matrix, and embryogenesis in Caenorhabditis elegans and Drosophila melanogaster. The results provide a visceral reminder of the beauty and the complexity of living systems.
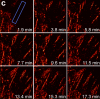
We demonstrate live-cell super-resolution imaging using photoactivated localization microscopy (PALM). The use of photon-tolerant cell lines in combination with the high resolution and molecular sensitivity of PALM permitted us to investigate the nanoscale dynamics within individual adhesion complexes (ACs) in living cells under physiological conditions for as long as 25 min, with half of the time spent collecting the PALM images at spatial resolutions down to approximately 60 nm and frame rates as short as 25 s. We visualized the formation of ACs and measured the fractional gain and loss of individual paxillin molecules as each AC evolved. By allowing observation of a wide variety of nanoscale dynamics, live-cell PALM provides insights into molecular assembly during the initiation, maturation and dissolution of cellular processes.
Commentary: The first example of true live cell and time lapse imaging by localization microscopy (as opposed to particle tracking), this paper uses the Nyquist criterion to establish a necessary condition for true spatial resolution based on the density of localized molecules – a condition often unmet in claims elsewhere in the superresolution literature.
By any method, higher spatiotemporal resolution requires increasing light exposure at the specimen, making noninvasive imaging increasingly difficult. Here, simultaneous differential interference contrast imaging is used to establish that cells behave physiologically before, during, and after PALM imaging. Similar controls are lacking from many supposed “live cell” superresolution demonstrations.
Membrane remodeling is an essential part for transfer of components to and from the cell surface and membrane-bound organelles, and for changes in cell shape, particularly critical during cell division. Earlier analyses, based on classical optical live-cell imaging, mostly restricted by technical necessity to the attached bottom surface, showed persistent formation of endocytic clathrin pits and vesicles during mitosis. Taking advantage of the resolution, speed, and non-invasive illumination of the newly developed lattice light sheet fluorescence microscope, we reexamined their assembly dynamics over the entire cell surface and showed that clathrin pits form at a lower rate during late mitosis. Full-cell imaging measurements of cell surface area and volume throughout the cell cycle of single cells in culture and in zebrafish embryos showed that the total surface increased rapidly during the transition from telophase to cytokinesis, whereas cell volume increased slightly in metaphase and remained relatively constant during cytokinesis. These applications demonstrate the advantage of lattice light sheet microscopy and enable a new standard for imaging membrane dynamics in single cells and in multicellular assemblies.
Optical nanoscopy of intact biological specimens has been transformed by recent advancements in hydrogel-based tissue clearing and expansion, enabling the imaging of cellular and subcellular structures with molecular contrast. However, existing high-resolution fluorescence microscopes are physically limited by objective-to-specimen distance, which prevents the study of whole-mount specimens without physical sectioning. To address this challenge, we developed a photochemical strategy for spatially precise sectioning of specimens. By combining serial photochemical sectioning with lattice light-sheet imaging and petabyte-scale computation, we imaged and reconstructed axons and myelin sheaths across entire mouse olfactory bulbs at nanoscale resolution. An olfactory bulb–wide analysis of myelinated and unmyelinated axons revealed distinctive patterns of axon degeneration and de-/dysmyelination in the neurodegenerative brain, highlighting the potential for peta- to exabyte-scale super-resolution studies using this approach. High-resolution microscopes have a short working distance, making it difficult to see deep within large biological samples such as an intact brain. Slicing the tissue with a blade can reach deeper, but this often distorts or destroys the fine structures that scientists want to study. By embedding a sample in a light-sensitive hydrogel, Wang et al. demonstrated a gentler approach using a precise ray or sheet of light to dissolve or cut away tissue layer by layer. After each layer is removed, the newly exposed surface is imaged, allowing for a complete, high-resolution, three-dimensional reconstruction without damaging physical contact. bioRxiv preprint: https://www.biorxiv.org/content/10.1101/2024.08.01.605857v1
Recent methods have revealed that cells on planar substrates exert both shear (in-plane) and normal (out-of-plane) tractions against the extracellular matrix (ECM). However, the location and origin of the normal tractions with respect to the adhesive and cytoskeletal elements of cells have not been elucidated. We developed a high-spatiotemporal-resolution, multidimensional (2.5D) traction force microscopy to measure and model the full 3D nature of cellular forces on planar 2D surfaces. We show that shear tractions are centered under elongated focal adhesions whereas upward and downward normal tractions are detected on distal (toward the cell edge) and proximal (toward the cell body) ends of adhesions, respectively. Together, these forces produce significant rotational moments about focal adhesions in both protruding and retracting peripheral regions. Temporal 2.5D traction force microscopy analysis of migrating and spreading cells shows that these rotational moments are highly dynamic, propagating outward with the leading edge of the cell. Finally, we developed a finite element model to examine how rotational moments could be generated about focal adhesions in a thin lamella. Our model suggests that rotational moments can be generated largely via shear lag transfer to the underlying ECM from actomyosin contractility applied at the intracellular surface of a rigid adhesion of finite thickness. Together, these data demonstrate and probe the origin of a previously unappreciated multidimensional stress profile associated with adhesions and highlight the importance of new approaches to characterize cellular forces.