Main Menu (Mobile)- Block
MENU
- Overview
-
Support Teams
- Overview
- Anatomy and Histology
- Cryo-Electron Microscopy
- Electron Microscopy
- Flow Cytometry
- Gene Targeting and Transgenics
- High Performance Computing
- Immortalized Cell Line Culture
- Integrative Imaging
- Invertebrate Shared Resource
- Janelia Experimental Technology
- Mass Spectrometry
- Media Prep
- Molecular Genomics
- Stem Cell & Primary Culture
- Project Pipeline Support
- Project Technical Resources
- Quantitative Genomics
- Scientific Computing
- Viral Tools
- Vivarium
- Open Science
- You + Janelia
- About Us
Labs:
Project Teams:
Main Menu - Block
Labs:
Project Teams:
- Overview
- Anatomy and Histology
- Cryo-Electron Microscopy
- Electron Microscopy
- Flow Cytometry
- Gene Targeting and Transgenics
- High Performance Computing
- Immortalized Cell Line Culture
- Integrative Imaging
- Invertebrate Shared Resource
- Janelia Experimental Technology
- Mass Spectrometry
- Media Prep
- Molecular Genomics
- Stem Cell & Primary Culture
- Project Pipeline Support
- Project Technical Resources
- Quantitative Genomics
- Scientific Computing
- Viral Tools
- Vivarium
node:field_image_thumbnail | entity_field
custom_misc-custom_misc_featured_summary | block
Harris Lab /
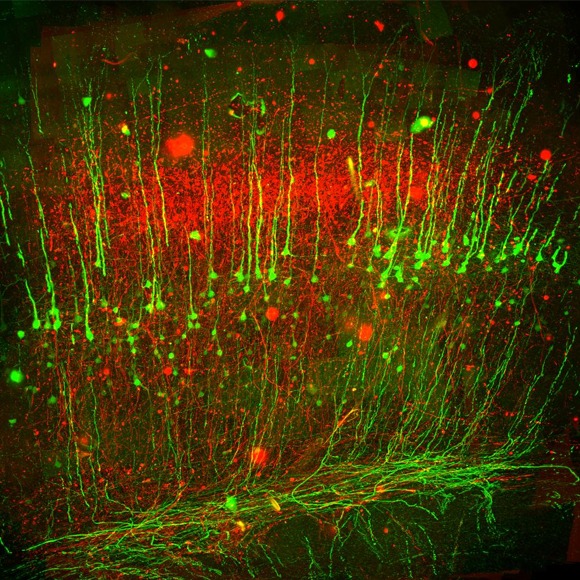
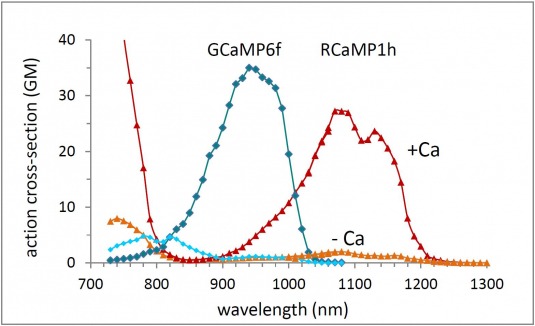
The Applied Physics & Instrumentation Group (APIG) develops unique tools to help Janelia researchers investigate the anatomy, activity, and connectivity of neurons and neural circuits.
janelia7_blocks-janelia7_secondary_menu | block
More in this Lab Landing Page
custom_misc-custom_misc_lab_updates | block
janelia7_blocks-janelia7_featured_blocks | block


node:field_content_summary | entity_field
APIG has a special role: Although a research lab, our goal is to create tools for other labs at Janelia. Coming from backgrounds in physics, chemistry, and engineering, we recruit, improve and invent next generation technologies for our Janelia neuroscience colleagues.
node:body | entity_field
node:field_pullquote_text | entity_field
Progress in science depends on new techniques, new discoveries and new ideas, probably in that order.
-Sydney Brenner