Filter
Associated Lab
- Aguilera Castrejon Lab (4) Apply Aguilera Castrejon Lab filter
- Ahrens Lab (64) Apply Ahrens Lab filter
- Aso Lab (42) Apply Aso Lab filter
- Baker Lab (19) Apply Baker Lab filter
- Betzig Lab (104) Apply Betzig Lab filter
- Beyene Lab (10) Apply Beyene Lab filter
- Bock Lab (14) Apply Bock Lab filter
- Branson Lab (53) Apply Branson Lab filter
- Card Lab (37) Apply Card Lab filter
- Cardona Lab (45) Apply Cardona Lab filter
- Chklovskii Lab (10) Apply Chklovskii Lab filter
- Clapham Lab (15) Apply Clapham Lab filter
- Cui Lab (19) Apply Cui Lab filter
- Darshan Lab (8) Apply Darshan Lab filter
- Dennis Lab (2) Apply Dennis Lab filter
- Dickson Lab (32) Apply Dickson Lab filter
- Druckmann Lab (21) Apply Druckmann Lab filter
- Dudman Lab (46) Apply Dudman Lab filter
- Eddy/Rivas Lab (30) Apply Eddy/Rivas Lab filter
- Egnor Lab (4) Apply Egnor Lab filter
- Espinosa Medina Lab (21) Apply Espinosa Medina Lab filter
- Feliciano Lab (16) Apply Feliciano Lab filter
- Fetter Lab (31) Apply Fetter Lab filter
- FIB-SEM Technology (1) Apply FIB-SEM Technology filter
- Fitzgerald Lab (17) Apply Fitzgerald Lab filter
- Freeman Lab (15) Apply Freeman Lab filter
- Funke Lab (46) Apply Funke Lab filter
- Gonen Lab (59) Apply Gonen Lab filter
- Grigorieff Lab (34) Apply Grigorieff Lab filter
- Harris Lab (55) Apply Harris Lab filter
- Heberlein Lab (13) Apply Heberlein Lab filter
- Hermundstad Lab (28) Apply Hermundstad Lab filter
- Hess Lab (77) Apply Hess Lab filter
- Ilanges Lab (4) Apply Ilanges Lab filter
- Jayaraman Lab (45) Apply Jayaraman Lab filter
- Ji Lab (33) Apply Ji Lab filter
- Johnson Lab (2) Apply Johnson Lab filter
- Karpova Lab (14) Apply Karpova Lab filter
- Keleman Lab (8) Apply Keleman Lab filter
- Keller Lab (62) Apply Keller Lab filter
- Koay Lab (4) Apply Koay Lab filter
- Lavis Lab (150) Apply Lavis Lab filter
- Lee (Albert) Lab (29) Apply Lee (Albert) Lab filter
- Leonardo Lab (19) Apply Leonardo Lab filter
- Li Lab (8) Apply Li Lab filter
- Lippincott-Schwartz Lab (110) Apply Lippincott-Schwartz Lab filter
- Liu (Yin) Lab (4) Apply Liu (Yin) Lab filter
- Liu (Zhe) Lab (60) Apply Liu (Zhe) Lab filter
- Looger Lab (137) Apply Looger Lab filter
- Magee Lab (31) Apply Magee Lab filter
- Menon Lab (12) Apply Menon Lab filter
- Murphy Lab (6) Apply Murphy Lab filter
- O'Shea Lab (7) Apply O'Shea Lab filter
- Otopalik Lab (1) Apply Otopalik Lab filter
- Pachitariu Lab (44) Apply Pachitariu Lab filter
- Pastalkova Lab (6) Apply Pastalkova Lab filter
- Pavlopoulos Lab (7) Apply Pavlopoulos Lab filter
- Pedram Lab (4) Apply Pedram Lab filter
- Podgorski Lab (16) Apply Podgorski Lab filter
- Reiser Lab (49) Apply Reiser Lab filter
- Riddiford Lab (20) Apply Riddiford Lab filter
- Romani Lab (40) Apply Romani Lab filter
- Rubin Lab (111) Apply Rubin Lab filter
- Saalfeld Lab (49) Apply Saalfeld Lab filter
- Satou Lab (3) Apply Satou Lab filter
- Scheffer Lab (38) Apply Scheffer Lab filter
- Schreiter Lab (55) Apply Schreiter Lab filter
- Schulze Lab (1) Apply Schulze Lab filter
- Sgro Lab (3) Apply Sgro Lab filter
- Shroff Lab (35) Apply Shroff Lab filter
- Simpson Lab (18) Apply Simpson Lab filter
- Singer Lab (37) Apply Singer Lab filter
- Spruston Lab (62) Apply Spruston Lab filter
- Stern Lab (77) Apply Stern Lab filter
- Sternson Lab (47) Apply Sternson Lab filter
- Stringer Lab (41) Apply Stringer Lab filter
- Svoboda Lab (132) Apply Svoboda Lab filter
- Tebo Lab (12) Apply Tebo Lab filter
- Tervo Lab (10) Apply Tervo Lab filter
- Tillberg Lab (19) Apply Tillberg Lab filter
- Tjian Lab (17) Apply Tjian Lab filter
- Truman Lab (58) Apply Truman Lab filter
- Turaga Lab (41) Apply Turaga Lab filter
- Turner Lab (27) Apply Turner Lab filter
- Vale Lab (8) Apply Vale Lab filter
- Voigts Lab (5) Apply Voigts Lab filter
- Wang (Meng) Lab (31) Apply Wang (Meng) Lab filter
- Wang (Shaohe) Lab (6) Apply Wang (Shaohe) Lab filter
- Wong-Campos Lab (1) Apply Wong-Campos Lab filter
- Wu Lab (8) Apply Wu Lab filter
- Zlatic Lab (26) Apply Zlatic Lab filter
- Zuker Lab (5) Apply Zuker Lab filter
Associated Project Team
- CellMap (13) Apply CellMap filter
- COSEM (3) Apply COSEM filter
- FIB-SEM Technology (5) Apply FIB-SEM Technology filter
- Fly Descending Interneuron (12) Apply Fly Descending Interneuron filter
- Fly Functional Connectome (14) Apply Fly Functional Connectome filter
- Fly Olympiad (5) Apply Fly Olympiad filter
- FlyEM (56) Apply FlyEM filter
- FlyLight (50) Apply FlyLight filter
- GENIE (47) Apply GENIE filter
- Integrative Imaging (11) Apply Integrative Imaging filter
- Larval Olympiad (2) Apply Larval Olympiad filter
- MouseLight (18) Apply MouseLight filter
- NeuroSeq (1) Apply NeuroSeq filter
- ThalamoSeq (1) Apply ThalamoSeq filter
- Tool Translation Team (T3) (29) Apply Tool Translation Team (T3) filter
- Transcription Imaging (45) Apply Transcription Imaging filter
Associated Support Team
- Project Pipeline Support (5) Apply Project Pipeline Support filter
- Anatomy and Histology (18) Apply Anatomy and Histology filter
- Cryo-Electron Microscopy (49) Apply Cryo-Electron Microscopy filter
- Electron Microscopy (18) Apply Electron Microscopy filter
- Gene Targeting and Transgenics (11) Apply Gene Targeting and Transgenics filter
- High Performance Computing (7) Apply High Performance Computing filter
- Integrative Imaging (25) Apply Integrative Imaging filter
- Invertebrate Shared Resource (40) Apply Invertebrate Shared Resource filter
- Janelia Experimental Technology (40) Apply Janelia Experimental Technology filter
- Management Team (1) Apply Management Team filter
- Mass Spectrometry (1) Apply Mass Spectrometry filter
- Molecular Genomics (15) Apply Molecular Genomics filter
- Project Technical Resources (54) Apply Project Technical Resources filter
- Quantitative Genomics (20) Apply Quantitative Genomics filter
- Scientific Computing (106) Apply Scientific Computing filter
- Stem Cell & Primary Culture (14) Apply Stem Cell & Primary Culture filter
- Viral Tools (14) Apply Viral Tools filter
- Vivarium (7) Apply Vivarium filter
Publication Date
- 2026 (118) Apply 2026 filter
- 2025 (223) Apply 2025 filter
- 2024 (208) Apply 2024 filter
- 2023 (157) Apply 2023 filter
- 2022 (166) Apply 2022 filter
- 2021 (175) Apply 2021 filter
- 2020 (177) Apply 2020 filter
- 2019 (177) Apply 2019 filter
- 2018 (206) Apply 2018 filter
- 2017 (186) Apply 2017 filter
- 2016 (191) Apply 2016 filter
- 2015 (195) Apply 2015 filter
- 2014 (190) Apply 2014 filter
- 2013 (136) Apply 2013 filter
- 2012 (112) Apply 2012 filter
- 2011 (98) Apply 2011 filter
- 2010 (61) Apply 2010 filter
- 2009 (56) Apply 2009 filter
- 2008 (40) Apply 2008 filter
- 2007 (21) Apply 2007 filter
- 2006 (3) Apply 2006 filter
2896 Janelia Publications
Showing 611-620 of 2896 resultsChemistry, once king of fluorescence microscopy, was usurped by the field of fluorescent proteins. The increased demands of modern microscopy techniques on the “photon budget” requires better and brighter fluorophores. Here, we review the recent advances in biochemistry, protein engineering, and organic synthesis that have allowed a triumphant return of chemical dyes to modern biological imaging.
The development of single-molecule localization microscopy (SMLM) has sparked a revolution in biological imaging, allowing 'super-resolution' fluorescence microscopy below the diffraction limit of light. The last decade has seen an explosion in not only optical hardware for SMLM but also the development or repurposing of fluorescent proteins and small-molecule fluorescent probes for this technique. In this review, written by chemists for chemists, we detail the history of single-molecule localization microscopy and collate the collection of probes with demonstrated utility in SMLM. We hope it will serve as a primer for probe choice in localization microscopy as well as an inspiration for the development of new fluorophores that enable imaging of biological samples with exquisite detail.
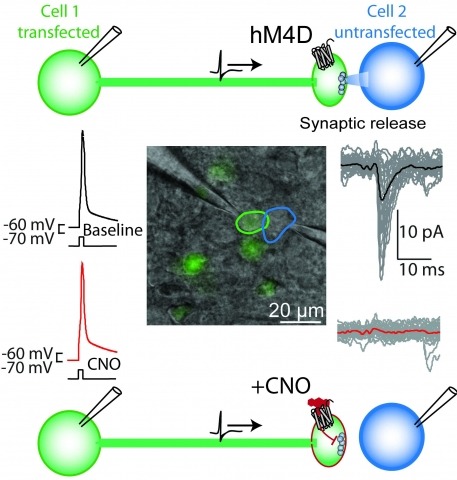
Brain function is mediated by neural circuit connectivity, and elucidating the role of connections is aided by techniques to block their output. We developed cell-type-selective, reversible synaptic inhibition tools for mammalian neural circuits by leveraging G protein signaling pathways to suppress synaptic vesicle release. Here, we find that the pharmacologically selective designer Gi-protein-coupled receptor hM4D is a presynaptic silencer in the presence of its cognate ligand clozapine-N-oxide (CNO). Activation of hM4D signaling sharply reduced synaptic release probability and synaptic current amplitude. To demonstrate the utility of this tool for neural circuit perturbations, we developed an axon-selective hM4D-neurexin variant and used spatially targeted intracranial CNO injections to localize circuit connections from the hypothalamus to the midbrain responsible for feeding behavior. This synaptic silencing approach is broadly applicable for cell-type-specific and axon projection-selective functional analysis of diverse neural circuits.
Chemogenetic technologies enable selective pharmacological control of specific cell populations. An increasing number of approaches have been developed that modulate different signaling pathways. Selective pharmacological control over G protein-coupled receptor signaling, ion channel conductances, protein association, protein stability, and small molecule targeting allows modulation of cellular processes in distinct cell types. Here, we review these chemogenetic technologies and instances of their applications in complex tissues in vivo and ex vivo.
Elucidating the roles of neuronal cell types for physiology and behavior is essential for understanding brain functions. Perturbation of neuron electrical activity can be used to probe the causal relationship between neuronal cell types and behavior. New genetically encoded neuron perturbation tools have been developed for remotely controlling neuron function using small molecules that activate engineered receptors that can be targeted to cell types using genetic methods. Here we describe recent progress for approaches using genetically engineered receptors that selectively interact with small molecules. Called "chemogenetics," receptors with diverse cellular functions have been developed that facilitate the selective pharmacological control over a diverse range of cell-signaling processes, including electrical activity, for molecularly defined cell types. These tools have revealed remarkably specific behavioral physiological influences for molecularly defined cell types that are often intermingled with populations having different or even opposite functions.
Chromatin immunoprecipitation (ChIP) is a technique that reveals in vivo location of a protein bound to DNA. ChIP coupled with DNA microarrays (ChIP-chip) or next-generation sequencing (ChIP-seq) allows for identification of binding sites of transcription factors on a global scale. Here we describe a protocol for ChIP to identify binding of the Ultrabithorax (Ubx) Hox transcription factors from imaginal discs of Drosophila larvae. The protocol can be extended to other model organisms and transcription factors.
Understanding how neurons integrate into developing circuits and contribute to functional activity is essential for decoding brain development and plasticity. However, current methods to study neuronal integration often suffer from low throughput, limited spatiotemporal resolution, or invasive procedures that hinder in vivo functional analysis. To overcome these challenges, we present a birthdate-labeling strategy, named CHLOK, based on HaloTag technology and a broad palette of fluorescent synthetic dyes. This approach enables precise multicolor labeling of neurons according to their maturation stage and allows flexible integration into functional assays through compatibility with calcium imaging and optogenetics. We validated CHLOK by mapping birthdate-resolved neuronal activity in the developing visual and motor systems of zebrafish larvae. Our results reveal distinct functional contributions of early- versus late-born neurons, providing new insights into the temporal dynamics of circuit formation. Furthermore, we demonstrate the versatility of this approach, showcasing age-specific multicolor calcium and voltage imaging as well as optogenetic manipulation. By overcoming key limitations of existing techniques, CHLOK offers a powerful, versatile and non-invasive tool for studying neural integration, circuit development and function in vivo.
Plastid isoprenoid-derived carotenoids serve essential roles in chloroplast development and photosynthesis. Although nearly all enzymes that participate in the biosynthesis of carotenoids in plants have been identified, the complement of auxiliary proteins that regulate synthesis, transport, sequestration, and degradation of these molecules and their isoprenoid precursors have not been fully described. To identify such proteins that are necessary for the optimal functioning of oxygenic photosynthesis, we screened a large collection of nonphotosynthetic (acetate-requiring) DNA insertional mutants of and isolated The mutant is extremely light-sensitive and susceptible to photoinhibition and photobleaching. The gene encodes a CRAL-TRIO hydrophobic ligand-binding (Sec14) domain protein. Proteins containing this domain are limited to eukaryotes, but some may have been retargeted to function in organelles of endosymbiotic origin. The mutant showed decreased accumulation of plastidial isoprenoid-derived pigments, especially carotenoids, and whole-cell focused ion-beam scanning-electron microscopy revealed a deficiency of carotenoid-rich chloroplast structures (e.g., eyespot and plastoglobules). The low carotenoid content resulted from impaired biosynthesis at a step prior to phytoene, the committed precursor to carotenoids. The CPSFL1 protein bound phytoene and β-carotene when expressed in and phosphatidic acid in vitro. We suggest that CPSFL1 is involved in the regulation of phytoene synthesis and carotenoid transport and thereby modulates carotenoid accumulation in the chloroplast.
Cells require a constant supply of fatty acids to survive and proliferate. Fatty acids incorporate into membrane and storage glycerolipids through a series of endoplasmic reticulum (ER) enzymes, but how these enzymes are regulated is not well understood. Here, using a combination of CRISPR-based genetic screens and unbiased lipidomics, we identified calcineurin B homologous protein 1 (CHP1) as a major regulator of ER glycerolipid synthesis. Loss of CHP1 severely reduces fatty acid incorporation and storage in mammalian cells and invertebrates. Mechanistically, CHP1 binds and activates GPAT4, which catalyzes the initial rate-limiting step in glycerolipid synthesis. GPAT4 activity requires CHP1 to be N-myristoylated, forming a key molecular interface between the two proteins. Interestingly, upon CHP1 loss, the peroxisomal enzyme, GNPAT, partially compensates for the loss of ER lipid synthesis, enabling cell proliferation. Thus, our work identifies a conserved regulator of glycerolipid metabolism and reveals plasticity in lipid synthesis of proliferating cells.
Three-dimensional (3D) chromatin organization plays a key role in regulating mammalian genome function; however, many of its physical features at the single-cell level remain underexplored. Here, we use live- and fixed-cell 3D super-resolution and scanning electron microscopy to analyze structural and functional nuclear organization in somatic cells. We identify chains of interlinked ~200- to 300-nm-wide chromatin domains (CDs) composed of aggregated nucleosomes that can overlap with individual topologically associating domains and are distinct from a surrounding RNA-populated interchromatin compartment. High-content mapping uncovers confinement of cohesin and active histone modifications to surfaces and enrichment of repressive modifications toward the core of CDs in both hetero- and euchromatic regions. This nanoscale functional topography is temporarily relaxed in postreplicative chromatin but remarkably persists after ablation of cohesin. Our findings establish CDs as physical and functional modules of mesoscale genome organization.