Filter
Associated Lab
- Aguilera Castrejon Lab (19) Apply Aguilera Castrejon Lab filter
- Ahrens Lab (75) Apply Ahrens Lab filter
- Aso Lab (42) Apply Aso Lab filter
- Baker Lab (38) Apply Baker Lab filter
- Betzig Lab (116) Apply Betzig Lab filter
- Beyene Lab (15) Apply Beyene Lab filter
- Bock Lab (17) Apply Bock Lab filter
- Branson Lab (56) Apply Branson Lab filter
- Card Lab (43) Apply Card Lab filter
- Cardona Lab (64) Apply Cardona Lab filter
- Chklovskii Lab (13) Apply Chklovskii Lab filter
- Clapham Lab (16) Apply Clapham Lab filter
- Cui Lab (19) Apply Cui Lab filter
- Darshan Lab (12) Apply Darshan Lab filter
- Dennis Lab (3) Apply Dennis Lab filter
- Dickson Lab (46) Apply Dickson Lab filter
- Druckmann Lab (25) Apply Druckmann Lab filter
- Dudman Lab (58) Apply Dudman Lab filter
- Eddy/Rivas Lab (30) Apply Eddy/Rivas Lab filter
- Egnor Lab (11) Apply Egnor Lab filter
- Espinosa Medina Lab (25) Apply Espinosa Medina Lab filter
- Feliciano Lab (16) Apply Feliciano Lab filter
- Fetter Lab (41) Apply Fetter Lab filter
- FIB-SEM Technology (1) Apply FIB-SEM Technology filter
- Fitzgerald Lab (30) Apply Fitzgerald Lab filter
- Freeman Lab (15) Apply Freeman Lab filter
- Funke Lab (46) Apply Funke Lab filter
- Gonen Lab (91) Apply Gonen Lab filter
- Grigorieff Lab (62) Apply Grigorieff Lab filter
- Harris Lab (65) Apply Harris Lab filter
- Heberlein Lab (94) Apply Heberlein Lab filter
- Hermundstad Lab (32) Apply Hermundstad Lab filter
- Hess Lab (80) Apply Hess Lab filter
- Ilanges Lab (4) Apply Ilanges Lab filter
- Jayaraman Lab (49) Apply Jayaraman Lab filter
- Ji Lab (33) Apply Ji Lab filter
- Johnson Lab (7) Apply Johnson Lab filter
- Kainmueller Lab (19) Apply Kainmueller Lab filter
- Karpova Lab (15) Apply Karpova Lab filter
- Keleman Lab (13) Apply Keleman Lab filter
- Keller Lab (77) Apply Keller Lab filter
- Koay Lab (20) Apply Koay Lab filter
- Lavis Lab (162) Apply Lavis Lab filter
- Lee (Albert) Lab (34) Apply Lee (Albert) Lab filter
- Leonardo Lab (23) Apply Leonardo Lab filter
- Li Lab (32) Apply Li Lab filter
- Lippincott-Schwartz Lab (182) Apply Lippincott-Schwartz Lab filter
- Liu (Yin) Lab (9) Apply Liu (Yin) Lab filter
- Liu (Zhe) Lab (65) Apply Liu (Zhe) Lab filter
- Looger Lab (138) Apply Looger Lab filter
- Magee Lab (49) Apply Magee Lab filter
- Menon Lab (18) Apply Menon Lab filter
- Murphy Lab (13) Apply Murphy Lab filter
- O'Shea Lab (8) Apply O'Shea Lab filter
- Otopalik Lab (13) Apply Otopalik Lab filter
- Pachitariu Lab (56) Apply Pachitariu Lab filter
- Pastalkova Lab (19) Apply Pastalkova Lab filter
- Pavlopoulos Lab (19) Apply Pavlopoulos Lab filter
- Pedram Lab (15) Apply Pedram Lab filter
- Podgorski Lab (16) Apply Podgorski Lab filter
- Reiser Lab (55) Apply Reiser Lab filter
- Riddiford Lab (44) Apply Riddiford Lab filter
- Romani Lab (52) Apply Romani Lab filter
- Rubin Lab (149) Apply Rubin Lab filter
- Saalfeld Lab (66) Apply Saalfeld Lab filter
- Satou Lab (18) Apply Satou Lab filter
- Scheffer Lab (38) Apply Scheffer Lab filter
- Schreiter Lab (72) Apply Schreiter Lab filter
- Schulze Lab (1) Apply Schulze Lab filter
- Sgro Lab (23) Apply Sgro Lab filter
- Shroff Lab (35) Apply Shroff Lab filter
- Simpson Lab (23) Apply Simpson Lab filter
- Singer Lab (80) Apply Singer Lab filter
- Spruston Lab (98) Apply Spruston Lab filter
- Stern Lab (160) Apply Stern Lab filter
- Sternson Lab (54) Apply Sternson Lab filter
- Stringer Lab (44) Apply Stringer Lab filter
- Svoboda Lab (136) Apply Svoboda Lab filter
- Tebo Lab (36) Apply Tebo Lab filter
- Tervo Lab (10) Apply Tervo Lab filter
- Tillberg Lab (22) Apply Tillberg Lab filter
- Tjian Lab (64) Apply Tjian Lab filter
- Truman Lab (88) Apply Truman Lab filter
- Turaga Lab (53) Apply Turaga Lab filter
- Turner Lab (38) Apply Turner Lab filter
- Vale Lab (8) Apply Vale Lab filter
- Voigts Lab (5) Apply Voigts Lab filter
- Wang (Meng) Lab (31) Apply Wang (Meng) Lab filter
- Wang (Shaohe) Lab (25) Apply Wang (Shaohe) Lab filter
- Wong-Campos Lab (1) Apply Wong-Campos Lab filter
- Wu Lab (9) Apply Wu Lab filter
- Zlatic Lab (28) Apply Zlatic Lab filter
- Zuker Lab (25) Apply Zuker Lab filter
Associated Project Team
- CellMap (13) Apply CellMap filter
- COSEM (3) Apply COSEM filter
- FIB-SEM Technology (5) Apply FIB-SEM Technology filter
- Fly Descending Interneuron (12) Apply Fly Descending Interneuron filter
- Fly Functional Connectome (14) Apply Fly Functional Connectome filter
- Fly Olympiad (5) Apply Fly Olympiad filter
- FlyEM (56) Apply FlyEM filter
- FlyLight (50) Apply FlyLight filter
- GENIE (47) Apply GENIE filter
- Integrative Imaging (11) Apply Integrative Imaging filter
- Larval Olympiad (2) Apply Larval Olympiad filter
- MouseLight (18) Apply MouseLight filter
- NeuroSeq (1) Apply NeuroSeq filter
- ThalamoSeq (1) Apply ThalamoSeq filter
- Tool Translation Team (T3) (29) Apply Tool Translation Team (T3) filter
- Transcription Imaging (49) Apply Transcription Imaging filter
Publication Date
- 2026 (118) Apply 2026 filter
- 2025 (223) Apply 2025 filter
- 2024 (209) Apply 2024 filter
- 2023 (158) Apply 2023 filter
- 2022 (192) Apply 2022 filter
- 2021 (194) Apply 2021 filter
- 2020 (196) Apply 2020 filter
- 2019 (202) Apply 2019 filter
- 2018 (232) Apply 2018 filter
- 2017 (217) Apply 2017 filter
- 2016 (209) Apply 2016 filter
- 2015 (252) Apply 2015 filter
- 2014 (236) Apply 2014 filter
- 2013 (194) Apply 2013 filter
- 2012 (190) Apply 2012 filter
- 2011 (190) Apply 2011 filter
- 2010 (161) Apply 2010 filter
- 2009 (158) Apply 2009 filter
- 2008 (140) Apply 2008 filter
- 2007 (106) Apply 2007 filter
- 2006 (92) Apply 2006 filter
- 2005 (67) Apply 2005 filter
- 2004 (57) Apply 2004 filter
- 2003 (58) Apply 2003 filter
- 2002 (39) Apply 2002 filter
- 2001 (28) Apply 2001 filter
- 2000 (29) Apply 2000 filter
- 1999 (14) Apply 1999 filter
- 1998 (18) Apply 1998 filter
- 1997 (16) Apply 1997 filter
- 1996 (10) Apply 1996 filter
- 1995 (18) Apply 1995 filter
- 1994 (12) Apply 1994 filter
- 1993 (10) Apply 1993 filter
- 1992 (6) Apply 1992 filter
- 1991 (11) Apply 1991 filter
- 1990 (11) Apply 1990 filter
- 1989 (6) Apply 1989 filter
- 1988 (1) Apply 1988 filter
- 1987 (7) Apply 1987 filter
- 1986 (4) Apply 1986 filter
- 1985 (5) Apply 1985 filter
- 1984 (2) Apply 1984 filter
- 1983 (2) Apply 1983 filter
- 1982 (3) Apply 1982 filter
- 1981 (3) Apply 1981 filter
- 1980 (1) Apply 1980 filter
- 1979 (1) Apply 1979 filter
- 1976 (2) Apply 1976 filter
- 1973 (1) Apply 1973 filter
- 1970 (1) Apply 1970 filter
- 1967 (1) Apply 1967 filter
Type of Publication
4313 Publications
Showing 2041-2050 of 4313 resultsBackground/Objectives: High-grade gliomas (HGGs), including glioblastomas, are among the most aggressive brain tumorså due to their high intratumoral heterogeneity and extensive infiltration. Glioma stem-like cells (GSCs) frequently invade along white matter tracts such as the corpus callosum, but the molecular programs driving this region-specific invasion remain poorly defined. The aim of this study was to identify transcriptional signatures associated with GSC infiltration into the corpus callosum. Methods: We established an orthotopic xenograft model by implanting fluorescently labeled human GSCs into nude mouse brains. Tumor growth and invasion patterns were assessed using tissue clearing, light-sheet fluorescence microscopy, and histological analyses. To characterize region-specific molecular profiles, we performed microfluidic-based single-cell RNA expression analysis of 48 invasion- and stemness-related genes in cells isolated from the tumor bulk (TB) and corpus callosum (CC). Results: By six weeks post-implantation, GSCs displayed marked tropism for the corpus callosum, with distinct infiltration patterns captured by three-dimensional imaging. Single-cell gene expression profiling revealed significant differences in 7 of the 48 genes (14.6%) between TB- and CC-derived GSCs. These genes—NES, CCND1, GUSB, NOTCH1, E2F1, EGFR, and TGFB1—collectively defined a “corpus callosum invasion signature” (CC-Iv). CC-derived cells showed a unimodal, high-expression profile of CC-Iv genes, whereas TB cells exhibited bimodal distributions, suggesting heterogeneous transcriptional states. Importantly, higher CC-Iv expression correlated with worse survival in patients with low-grade gliomas. Conclusions: This multimodal approach identified a corpus callosum-specific invasion signature in glioma stem-like cells, revealing how local microenvironmental cues shape transcriptional reprogramming during infiltration. These findings provide new insights into the spatial heterogeneity of gliomas and highlight potential molecular targets for therapies designed to limit tumor spread through white matter tracts.
Deep neural networks trained to inpaint partially occluded images show a deep understanding of image composition and have even been shown to remove objects from images convincingly. In this work, we investigate how this implicit knowledge of image composition can be be used to separate cells in densely populated microscopy images. We propose a measure for the independence of two image regions given a fully self-supervised inpainting network and separate objects by maximizing this independence. We evaluate our method on two cell segmentation datasets and show that cells can be separated completely unsupervised. Furthermore, combined with simple foreground detection, our method yields instance segmentation of similar quality to fully supervised methods.
Different types of Drosophila dopaminergic neurons (DANs) reinforce memories of unique valence and provide state-dependent motivational control [1]. Prior studies suggest that the compartment architecture of the mushroom body (MB) is the relevant resolution for distinct DAN functions [2, 3]. Here we used a recent electron microscope volume of the fly brain [4] to reconstruct the fine anatomy of individual DANs within three MB compartments. We find the 20 DANs of the γ5 compartment, at least some of which provide reward teaching signals, can be clustered into 5 anatomical subtypes that innervate different regions within γ5. Reconstructing 821 upstream neurons reveals input selectivity, supporting the functional relevance of DAN sub-classification. Only one PAM-γ5 DAN subtype (γ5fb) receives direct recurrent input from γ5β’2a mushroom body output neurons (MBONs) and behavioral experiments distinguish a role for these DANs in memory revaluation from those reinforcing sugar memory. Other DAN subtypes receive major, and potentially reinforcing, inputs from putative gustatory interneurons or lateral horn neurons, which can also relay indirect feedback from the MB. We similarly reconstructed the single aversively reinforcing PPL1-γ1pedc DAN. The γ1pedc DAN inputs are mostly different to those of γ5 DANs and are clustered onto distinct branches of its dendritic tree, presumably separating its established roles in aversive reinforcement and appetitive motivation [5, 6]. Additional tracing identified neurons that provide broad input to γ5, β’2a and γ1pedc DANs suggesting that distributed DAN populations can be coordinately regulated. These connectomic and behavioral analyses therefore reveal additional complexity of dopaminergic reinforcement circuits between and within MB compartments.
The ability to discriminate sensory stimuli with overlapping features is thought to arise in brain structures called expansion layers, where neurons carrying information about sensory features make combinatorial connections onto a much larger set of cells. For 50 years, expansion coding has been a prime topic of theoretical neuroscience, which seeks to explain how quantitative parameters of the expansion circuit influence sensory sensitivity, discrimination, and generalization. Here, we investigate the developmental events that produce the quantitative parameters of the arthropod expansion layer, called the mushroom body. Using Drosophila melanogaster as a model, we employ genetic and chemical tools to engineer changes to circuit development. These allow us to produce living animals with hypothesis-driven variations on natural expansion layer wiring parameters. We then test the functional and behavioral consequences. By altering the number of expansion layer neurons (Kenyon cells) and their dendritic complexity, we find that input density, but not cell number, tunes neuronal odor selectivity. Simple odor discrimination behavior is maintained when the Kenyon cell number is reduced and augmented by Kenyon cell number expansion. Animals with increased input density to each Kenyon cell show increased overlap in Kenyon cell odor responses and become worse at odor discrimination tasks.
The ability to discriminate sensory stimuli with overlapping features is thought to arise in brain structures called expansion layers, where neurons carrying information about sensory features make combinatorial connections onto a much larger set of cells. For 50 years, expansion coding has been a prime topic of theoretical neuroscience, which seeks to explain how quantitative parameters of the expansion circuit influence sensory sensitivity, discrimination, and generalization. Here, we investigate the developmental events that produce the quantitative parameters of the arthropod expansion layer, called the mushroom body. Using Drosophila melanogaster as a model, we employ genetic and chemical tools to engineer changes to circuit development. These allow us to produce living animals with hypothesis-driven variations on natural expansion layer wiring parameters. We then test the functional and behavioral consequences. By altering the number of expansion layer neurons (Kenyon cells) and their dendritic complexity, we find that input density, but not cell number, tunes neuronal odor selectivity. Simple odor discrimination behavior is maintained when the Kenyon cell number is reduced and augmented by Kenyon cell number expansion. Animals with increased input density to each Kenyon cell show increased overlap in Kenyon cell odor responses and become worse at odor discrimination tasks.
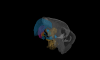
The basal ganglia play a critical role in the regulation of voluntary action in vertebrates. Our understanding of the function of the basal ganglia relies heavily upon anatomical information, but continued progress will require an understanding of the specific functional roles played by diverse cell types and their connectivity. An increasing number of mouse lines allow extensive identification, characterization, and manipulation of specified cell types in the basal ganglia. Despite the promise of genetically modified mice for elucidating the functional roles of diverse cell types, there is relatively little anatomical data obtained directly in the mouse. Here we have characterized the retrograde labeling obtained from a series of tracer injections throughout the dorsal striatum of adult mice. We found systematic variations in input along both the medial-lateral and anterior-posterior neuraxes in close agreement with canonical features of basal ganglia anatomy in the rat. In addition to the canonical features we have provided experimental support for the importance of non-canonical inputs to the striatum from the raphe nuclei and the amygdala. To look for organization at a finer scale we have analyzed the correlation structure of labeling intensity across our entire dataset. Using this analysis we found substantial local heterogeneity within the large-scale order. From this analysis we conclude that individual striatal sites receive varied combinations of cortical and thalamic input from multiple functional areas, consistent with some earlier studies in the rat that have suggested the presence of a combinatorial map.
It is known that during lens differentiation a number of fibre cell specific membrane proteins change their expression profiles. In this study we have investigated how the profiles of the two most abundant fibre cell membrane proteins AQP0 (formerly known as Major Intrinsic Protein, MIP) and MP20 change as a function of fibre cell differentiation. While AQP0 was always found associated with fibre cell membranes, MP20 was initially found in the cytoplasm of peripheral fibre cells before becoming inserted into the membranes of deeper fibre cells. To determine at what stage in fibre cell differentiation MP20 becomes inserted into the membrane, sections were double-labelled with an antibody against MP20, and propidium iodide, a marker of cell nuclei. This showed that membrane insertion of MP20 occurs in a discrete transition zone that coincided with the degradation of cell nuclei. To test the significance of the membrane insertion of MP20 to overall lens function, whole lenses were incubated for varying times in a solution containing either Texas Red-dextran or Lucifer yellow as markers of extracellular space. Lenses were fixed and then processed for immunocytochemistry. Analysis of these sections showed that both tracer dyes were excluded from the extracellular space in an area that coincided with insertion of MP20 into the plasma membrane. Our results suggest that the insertion of MP20 into fibre cell membranes coincides with the creation of a barrier that restricts the diffusion of molecules into the lens core via the extracellular space.
Fruit flies (Drosophila melanogaster) are small insects, with correspondingly small power budgets. Despite this, they perform sophisticated neural computations in real time. Careful study of these insects is revealing how some of these circuits work. Insights from these systems might be helpful in designing other low power circuits.
AIM: Ethanol-induced locomotor sensitization is a behavioral manifestation of physiological responses to repeated ethanol exposures. While ethanol exerts direct effects on multiple neurotransmitter systems in the brain, ethanol-induced changes in metabolic state, including acute hyperglycemia and inhibition of insulin signaling, also have plausible roles in the expression of ethanol-related behaviors through direct and indirect effects on brain function. The current experiments examined whether insulin administration or the resultant hypoglycemia might attenuate the development of sensitization to the locomotor stimulant effect of ethanol. MAIN METHODS: Male and female DBA/2J mice received daily injections of 5 or 10 IU/kg insulin before or after a stimulating dose of ethanol and subsequent testing in an automated activity monitor. Blood glucose levels were determined upon the completion of the experiments. KEY FINDINGS: Insulin injected prior to ethanol blunted the acute stimulant response as well as the acquisition and expression of locomotor sensitization, while insulin given after ethanol did not affect the development of the sensitized response. In a separate experiment, mice given glucose concurrently with insulin developed ethanol-induced locomotor sensitization normally. SIGNIFICANCE: These experiments suggest that insulin attenuates the development of ethanol-induced locomotor sensitization, and that blood glucose levels can largely account for this effect. Further studies of the role of ethanol-induced metabolic states should provide novel information on the expression of ethanol-related behaviors.
The insulin signaling pathway regulates multiple physiological processes, including energy metabolism, organismal growth, aging and reproduction. Here we show that genetic manipulations in Drosophila melanogaster that impair the function of insulin-producing cells or of the insulin-receptor signaling pathway in the nervous system lead to increased sensitivity to the intoxicating effects of ethanol. These findings suggest a previously unknown role for this highly conserved pathway in regulating the behavioral responses to an addictive drug.