Filter
Associated Lab
- Aguilera Castrejon Lab (4) Apply Aguilera Castrejon Lab filter
- Ahrens Lab (64) Apply Ahrens Lab filter
- Aso Lab (42) Apply Aso Lab filter
- Baker Lab (19) Apply Baker Lab filter
- Betzig Lab (104) Apply Betzig Lab filter
- Beyene Lab (10) Apply Beyene Lab filter
- Bock Lab (14) Apply Bock Lab filter
- Branson Lab (53) Apply Branson Lab filter
- Card Lab (37) Apply Card Lab filter
- Cardona Lab (45) Apply Cardona Lab filter
- Chklovskii Lab (10) Apply Chklovskii Lab filter
- Clapham Lab (15) Apply Clapham Lab filter
- Cui Lab (19) Apply Cui Lab filter
- Darshan Lab (8) Apply Darshan Lab filter
- Dennis Lab (2) Apply Dennis Lab filter
- Dickson Lab (32) Apply Dickson Lab filter
- Druckmann Lab (21) Apply Druckmann Lab filter
- Dudman Lab (46) Apply Dudman Lab filter
- Eddy/Rivas Lab (30) Apply Eddy/Rivas Lab filter
- Egnor Lab (4) Apply Egnor Lab filter
- Espinosa Medina Lab (21) Apply Espinosa Medina Lab filter
- Feliciano Lab (16) Apply Feliciano Lab filter
- Fetter Lab (31) Apply Fetter Lab filter
- FIB-SEM Technology (1) Apply FIB-SEM Technology filter
- Fitzgerald Lab (17) Apply Fitzgerald Lab filter
- Freeman Lab (15) Apply Freeman Lab filter
- Funke Lab (46) Apply Funke Lab filter
- Gonen Lab (59) Apply Gonen Lab filter
- Grigorieff Lab (34) Apply Grigorieff Lab filter
- Harris Lab (55) Apply Harris Lab filter
- Heberlein Lab (13) Apply Heberlein Lab filter
- Hermundstad Lab (28) Apply Hermundstad Lab filter
- Hess Lab (77) Apply Hess Lab filter
- Ilanges Lab (4) Apply Ilanges Lab filter
- Jayaraman Lab (45) Apply Jayaraman Lab filter
- Ji Lab (33) Apply Ji Lab filter
- Johnson Lab (2) Apply Johnson Lab filter
- Karpova Lab (14) Apply Karpova Lab filter
- Keleman Lab (8) Apply Keleman Lab filter
- Keller Lab (62) Apply Keller Lab filter
- Koay Lab (4) Apply Koay Lab filter
- Lavis Lab (150) Apply Lavis Lab filter
- Lee (Albert) Lab (29) Apply Lee (Albert) Lab filter
- Leonardo Lab (19) Apply Leonardo Lab filter
- Li Lab (8) Apply Li Lab filter
- Lippincott-Schwartz Lab (110) Apply Lippincott-Schwartz Lab filter
- Liu (Yin) Lab (4) Apply Liu (Yin) Lab filter
- Liu (Zhe) Lab (60) Apply Liu (Zhe) Lab filter
- Looger Lab (137) Apply Looger Lab filter
- Magee Lab (31) Apply Magee Lab filter
- Menon Lab (12) Apply Menon Lab filter
- Murphy Lab (6) Apply Murphy Lab filter
- O'Shea Lab (7) Apply O'Shea Lab filter
- Otopalik Lab (1) Apply Otopalik Lab filter
- Pachitariu Lab (44) Apply Pachitariu Lab filter
- Pastalkova Lab (6) Apply Pastalkova Lab filter
- Pavlopoulos Lab (7) Apply Pavlopoulos Lab filter
- Pedram Lab (4) Apply Pedram Lab filter
- Podgorski Lab (16) Apply Podgorski Lab filter
- Reiser Lab (49) Apply Reiser Lab filter
- Riddiford Lab (20) Apply Riddiford Lab filter
- Romani Lab (40) Apply Romani Lab filter
- Rubin Lab (111) Apply Rubin Lab filter
- Saalfeld Lab (49) Apply Saalfeld Lab filter
- Satou Lab (3) Apply Satou Lab filter
- Scheffer Lab (38) Apply Scheffer Lab filter
- Schreiter Lab (55) Apply Schreiter Lab filter
- Schulze Lab (1) Apply Schulze Lab filter
- Sgro Lab (3) Apply Sgro Lab filter
- Shroff Lab (35) Apply Shroff Lab filter
- Simpson Lab (18) Apply Simpson Lab filter
- Singer Lab (37) Apply Singer Lab filter
- Spruston Lab (62) Apply Spruston Lab filter
- Stern Lab (77) Apply Stern Lab filter
- Sternson Lab (47) Apply Sternson Lab filter
- Stringer Lab (41) Apply Stringer Lab filter
- Svoboda Lab (132) Apply Svoboda Lab filter
- Tebo Lab (12) Apply Tebo Lab filter
- Tervo Lab (10) Apply Tervo Lab filter
- Tillberg Lab (19) Apply Tillberg Lab filter
- Tjian Lab (17) Apply Tjian Lab filter
- Truman Lab (58) Apply Truman Lab filter
- Turaga Lab (41) Apply Turaga Lab filter
- Turner Lab (27) Apply Turner Lab filter
- Vale Lab (8) Apply Vale Lab filter
- Voigts Lab (5) Apply Voigts Lab filter
- Wang (Meng) Lab (31) Apply Wang (Meng) Lab filter
- Wang (Shaohe) Lab (6) Apply Wang (Shaohe) Lab filter
- Wong-Campos Lab (1) Apply Wong-Campos Lab filter
- Wu Lab (8) Apply Wu Lab filter
- Zlatic Lab (26) Apply Zlatic Lab filter
- Zuker Lab (5) Apply Zuker Lab filter
Associated Project Team
- CellMap (13) Apply CellMap filter
- COSEM (3) Apply COSEM filter
- FIB-SEM Technology (5) Apply FIB-SEM Technology filter
- Fly Descending Interneuron (12) Apply Fly Descending Interneuron filter
- Fly Functional Connectome (14) Apply Fly Functional Connectome filter
- Fly Olympiad (5) Apply Fly Olympiad filter
- FlyEM (56) Apply FlyEM filter
- FlyLight (50) Apply FlyLight filter
- GENIE (47) Apply GENIE filter
- Integrative Imaging (11) Apply Integrative Imaging filter
- Larval Olympiad (2) Apply Larval Olympiad filter
- MouseLight (18) Apply MouseLight filter
- NeuroSeq (1) Apply NeuroSeq filter
- ThalamoSeq (1) Apply ThalamoSeq filter
- Tool Translation Team (T3) (29) Apply Tool Translation Team (T3) filter
- Transcription Imaging (45) Apply Transcription Imaging filter
Associated Support Team
- Project Pipeline Support (5) Apply Project Pipeline Support filter
- Anatomy and Histology (18) Apply Anatomy and Histology filter
- Cryo-Electron Microscopy (49) Apply Cryo-Electron Microscopy filter
- Electron Microscopy (18) Apply Electron Microscopy filter
- Gene Targeting and Transgenics (11) Apply Gene Targeting and Transgenics filter
- High Performance Computing (7) Apply High Performance Computing filter
- Integrative Imaging (25) Apply Integrative Imaging filter
- Invertebrate Shared Resource (40) Apply Invertebrate Shared Resource filter
- Janelia Experimental Technology (40) Apply Janelia Experimental Technology filter
- Management Team (1) Apply Management Team filter
- Mass Spectrometry (1) Apply Mass Spectrometry filter
- Molecular Genomics (15) Apply Molecular Genomics filter
- Project Technical Resources (54) Apply Project Technical Resources filter
- Quantitative Genomics (20) Apply Quantitative Genomics filter
- Scientific Computing (106) Apply Scientific Computing filter
- Stem Cell & Primary Culture (14) Apply Stem Cell & Primary Culture filter
- Viral Tools (14) Apply Viral Tools filter
- Vivarium (7) Apply Vivarium filter
Publication Date
- 2026 (118) Apply 2026 filter
- 2025 (223) Apply 2025 filter
- 2024 (208) Apply 2024 filter
- 2023 (157) Apply 2023 filter
- 2022 (166) Apply 2022 filter
- 2021 (175) Apply 2021 filter
- 2020 (177) Apply 2020 filter
- 2019 (177) Apply 2019 filter
- 2018 (206) Apply 2018 filter
- 2017 (186) Apply 2017 filter
- 2016 (191) Apply 2016 filter
- 2015 (195) Apply 2015 filter
- 2014 (190) Apply 2014 filter
- 2013 (136) Apply 2013 filter
- 2012 (112) Apply 2012 filter
- 2011 (98) Apply 2011 filter
- 2010 (61) Apply 2010 filter
- 2009 (56) Apply 2009 filter
- 2008 (40) Apply 2008 filter
- 2007 (21) Apply 2007 filter
- 2006 (3) Apply 2006 filter
2896 Janelia Publications
Showing 1551-1560 of 2896 resultsThe proliferation of microscopy methods for live-cell imaging offers many new possibilities for users but can also be challenging to navigate. The prevailing challenge in live-cell fluorescence microscopy is capturing intra-cellular dynamics while preserving cell viability. Computational methods can help to address this challenge and are now shifting the boundaries of what is possible to capture in living systems. In this Review, we discuss these computational methods focusing on artificial intelligence-based approaches that can be layered on top of commonly used existing microscopies as well as hybrid methods that integrate computation and microscope hardware. We specifically discuss how computational approaches can improve the signal-to-noise ratio, spatial resolution, temporal resolution and multi-colour capacity of live-cell imaging.
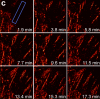
We demonstrate live-cell super-resolution imaging using photoactivated localization microscopy (PALM). The use of photon-tolerant cell lines in combination with the high resolution and molecular sensitivity of PALM permitted us to investigate the nanoscale dynamics within individual adhesion complexes (ACs) in living cells under physiological conditions for as long as 25 min, with half of the time spent collecting the PALM images at spatial resolutions down to approximately 60 nm and frame rates as short as 25 s. We visualized the formation of ACs and measured the fractional gain and loss of individual paxillin molecules as each AC evolved. By allowing observation of a wide variety of nanoscale dynamics, live-cell PALM provides insights into molecular assembly during the initiation, maturation and dissolution of cellular processes.
Commentary: The first example of true live cell and time lapse imaging by localization microscopy (as opposed to particle tracking), this paper uses the Nyquist criterion to establish a necessary condition for true spatial resolution based on the density of localized molecules – a condition often unmet in claims elsewhere in the superresolution literature.
By any method, higher spatiotemporal resolution requires increasing light exposure at the specimen, making noninvasive imaging increasingly difficult. Here, simultaneous differential interference contrast imaging is used to establish that cells behave physiologically before, during, and after PALM imaging. Similar controls are lacking from many supposed “live cell” superresolution demonstrations.
The H2A.Z histone variant, a genome-wide hallmark of permissive chromatin, is enriched near transcription start sites in all eukaryotes. H2A.Z is deposited by the SWR1 chromatin remodeler and evicted by unclear mechanisms. We tracked H2A.Z in living yeast at single-molecule resolution, and found that H2A.Z eviction is dependent on RNA Polymerase II (Pol II) and the Kin28/Cdk7 kinase, which phosphorylates Serine 5 of heptapeptide repeats on the carboxy-terminal domain of the largest Pol II subunit Rpb1. These findings link H2A.Z eviction to transcription initiation, promoter escape and early elongation activities of Pol II. Because passage of Pol II through +1 nucleosomes genome-wide would obligate H2A.Z turnover, we propose that global transcription at yeast promoters is responsible for eviction of H2A.Z. Such usage of yeast Pol II suggests a general mechanism coupling eukaryotic transcription to erasure of the H2A.Z epigenetic signal.
Stereocilia are F-actin-based cylindrical protrusions on the apical surface of inner ear hair cells that function as biological mechanosensors of sound and acceleration. During stereocilia development, specific unconventional myosins transport proteins and phospholipids as cargo and mediate elongation, differentiation and acquisition of the mechanoelectrical transduction (MET). How unconventional myosins localize themselves and cargo in stereocilia using energy from ATP hydrolysis is only partially understood. Here, we developed STELLA-SPIM microscopy to visualize movement of single myosin molecules in live hair cell stereocilia. STELLA-SPIM demonstrated that MYO7A, a component of MET machinery, shows processive movement toward stereocilia tips when chemically dimerized or constitutively activated by missense mutations disabling tail-mediated autoinhibition. Conversely, MYO7A shows step-wise but not processive movement in stereocilia when its tail is tethered to the plasma membrane or F-actin in the presence of MYO7A interacting partners. We posit that MYO7A dimerizes and moves processively in stereocilia when unleashed from autoinhibition.
The Polycomb PRC1 plays essential roles in development and disease pathogenesis. Targeting of PRC1 to chromatin is thought to be mediated by the Cbx family proteins (Cbx2/4/6/7/8) binding to histone H3 with a K27me3 modification (H3K27me3). Despite this prevailing view, the molecular mechanisms of targeting remain poorly understood. Here, by combining live-cell single-molecule tracking (SMT) and genetic engineering, we reveal that H3K27me3 contributes significantly to the targeting of Cbx7 and Cbx8 to chromatin, but less to Cbx2, Cbx4, and Cbx6. Genetic disruption of the complex formation of PRC1 facilitates the targeting of Cbx7 to chromatin. Biochemical analyses uncover that the CD and AT-hook-like (ATL) motif of Cbx7 constitute a functional DNA-binding unit. Live-cell SMT of Cbx7 mutants demonstrates that Cbx7 is targeted to chromatin by co-recognizing of H3K27me3 and DNA. Our data suggest a novel hierarchical cooperation mechanism by which histone modifications and DNA coordinate to target chromatin regulatory complexes.
The cortical actin cytoskeleton has been shown to be critical for the reorganization and heterogeneity of plasma membrane components of many cells, including T cells. Building on previous studies at the T cell immunological synapse, we quantitatively assess the structure and dynamics of this meshwork using live-cell superresolution fluorescence microscopy and spatio-temporal image correlation spectroscopy. We show for the first time, to our knowledge, that not only does the dense actin cortex flow in a retrograde fashion toward the synapse center, but the plasma membrane itself shows similar behavior. Furthermore, using two-color, live-cell superresolution cross-correlation spectroscopy, we demonstrate that the two flows are correlated and, in addition, we show that coupling may extend to the outer leaflet of the plasma membrane by examining the flow of GPI-anchored proteins. Finally, we demonstrate that the actin flow is correlated with a third component, α-actinin, which upon CRISPR knockout led to reduced plasma membrane flow directionality despite increased actin flow velocity. We hypothesize that this apparent cytoskeletal-membrane coupling could provide a mechanism for driving the observed retrograde flow of signaling molecules such as the TCR, Lck, ZAP70, LAT, and SLP76.
The second messenger cyclic AMP (cAMP) operates in discrete subcellular regions within which proteins that synthesize, break down or respond to the second messenger are precisely organized. A burgeoning knowledge of compartmentalized cAMP signaling is revealing how the local control of signaling enzyme activity impacts upon disease. The aim of this Cell Science at a Glance article and the accompanying poster is to highlight how misregulation of local cyclic AMP signaling can have pathophysiological consequences. We first introduce the core molecular machinery for cAMP signaling, which includes the cAMP-dependent protein kinase (PKA), and then consider the role of A-kinase anchoring proteins (AKAPs) in coordinating different cAMP-responsive proteins. The latter sections illustrate the emerging role of local cAMP signaling in four disease areas: cataracts, cancer, diabetes and cardiovascular diseases.
Accuracy of automated structural RNA alignment is improved by using models that consider not only primary sequence but also secondary structure information. However, current RNA structural alignment approaches tend to perform poorly on incomplete sequence fragments, such as single reads from metagenomic environmental surveys, because nucleotides that are expected to be base paired are missing.
We present a simple, yet effective, auxiliary learning task for the problem of neuron segmentation in electron microscopy volumes. The auxiliary task consists of the prediction of Local Shape Descriptors (LSDs), which we combine with conventional voxel-wise direct neighbor affinities for neuron boundary detection. The shape descriptors are designed to capture local statistics about the neuron to be segmented, such as diameter, elongation, and direction. On a large study comparing several existing methods across various specimen, imaging techniques, and resolutions, we find that auxiliary learning of LSDs consistently increases segmentation accuracy of affinity-based methods over a range of metrics. Furthermore, the addition of LSDs promotes affinitybased segmentation methods to be on par with the current state of the art for neuron segmentation (Flood-Filling Networks, FFN), while being two orders of magnitudes more efficient—a critical requirement for the processing of future petabyte-sized datasets. Implementations of the new auxiliary learning task, network architectures, training, prediction, and evaluation code, as well as the datasets used in this study are publicly available as a benchmark for future method contributions.Competing Interest StatementThe authors have declared no competing interest.
We present an auxiliary learning task for the problem of neuron segmentation in electron microscopy volumes. The auxiliary task consists of the prediction of local shape descriptors (LSDs), which we combine with conventional voxel-wise direct neighbor affinities for neuron boundary detection. The shape descriptors capture local statistics about the neuron to be segmented, such as diameter, elongation, and direction. On a study comparing several existing methods across various specimen, imaging techniques, and resolutions, auxiliary learning of LSDs consistently increases segmentation accuracy of affinity-based methods over a range of metrics. Furthermore, the addition of LSDs promotes affinity-based segmentation methods to be on par with the current state of the art for neuron segmentation (flood-filling networks), while being two orders of magnitudes more efficient-a critical requirement for the processing of future petabyte-sized datasets.