Filter
Associated Lab
- Betzig Lab (1) Apply Betzig Lab filter
- Dickson Lab (1) Apply Dickson Lab filter
- Harris Lab (2) Apply Harris Lab filter
- Lavis Lab (7) Apply Lavis Lab filter
- Lippincott-Schwartz Lab (1) Apply Lippincott-Schwartz Lab filter
- Liu (Zhe) Lab (2) Apply Liu (Zhe) Lab filter
- Rubin Lab (1) Apply Rubin Lab filter
- Saalfeld Lab (1) Apply Saalfeld Lab filter
- Remove Singer Lab filter Singer Lab
- Stern Lab (2) Apply Stern Lab filter
- Tjian Lab (5) Apply Tjian Lab filter
- Truman Lab (1) Apply Truman Lab filter
Associated Project Team
Associated Support Team
Publication Date
Tool Types
39 Results
Showing 1-10 of 39 resultsSpecific labeling of biomolecules with bright fluorophores is the keystone of fluorescence microscopy. Genetically encoded self-labeling tag proteins can be coupled to synthetic dyes inside living cells, resulting in brighter reporters than fluorescent proteins. Intracellular labeling using these techniques requires cell-permeable fluorescent ligands, however, limiting utility to a small number of classic fluorophores. Here we describe a simple structural modification that improves the brightness and photostability of dyes while preserving spectral properties and cell permeability. Inspired by molecular modeling, we replaced the N,N-dimethylamino substituents in tetramethylrhodamine with four-membered azetidine rings. This addition of two carbon atoms doubles the quantum efficiency and improves the photon yield of the dye in applications ranging from in vitro single-molecule measurements to super-resolution imaging. The novel substitution is generalizable, yielding a palette of chemical dyes with improved quantum efficiencies that spans the UV and visible range.
Spontaneously blinking fluorophores permit the detection and localization of individual molecules without reducing buffers or caging groups, thus simplifying single-molecule localization microscopy (SMLM). The intrinsic blinking properties of such dyes are dictated by molecular structure and modulated by environment, which can limit utility. We report a series of tuned spontaneously blinking dyes with duty cycles that span two orders of magnitude, allowing facile SMLM in cells and dense biomolecular structures.
Our aim is to develop quantitative single-molecule assays to study when and where molecules are interacting inside living cells and where enzymes are active. To this end we present a three-camera imaging microscope for fast tracking of multiple interacting molecules simultaneously, with high spatiotemporal resolution. The system was designed around an ASI RAMM frame using three separate tube lenses and custom multi-band dichroics to allow for enhanced detection efficiency. The frame times of the three Andor iXon Ultra EMCCD cameras are hardware synchronized to the laser excitation pulses of the three excitation lasers, such that the fluorophores are effectively immobilized during frame acquisitions and do not yield detections that are motion-blurred. Stroboscopic illumination allows robust detection from even rapidly moving molecules while minimizing bleaching, and since snapshots can be spaced out with varying time intervals, stroboscopic illumination enables a direct comparison to be made between fast and slow molecules under identical light dosage. We have developed algorithms that accurately track and co-localize multiple interacting biomolecules. The three-color microscope combined with our co-movement algorithms have made it possible for instance to simultaneously image and track how the chromosome environment affects diffusion kinetics or determine how mRNAs diffuse during translation. Such multiplexed single-molecule measurements at a high spatiotemporal resolution inside living cells will provide a major tool for testing models relating molecular architecture and biological dynamics.
Localized translation plays a crucial role in synaptic plasticity and memory consolidation. However, it has not been possible to follow the dynamics of memory-associated mRNAs in living neurons in response to neuronal activity in real time. We have generated a novel mouse model where the endogenous Arc/Arg3.1 gene is tagged in its 3' untranslated region with stem-loops that bind a bacteriophage PP7 coat protein (PCP), allowing visualization of individual mRNAs in real time. The physiological response of the tagged gene to neuronal activity is identical to endogenous Arc and reports the true dynamics of Arc mRNA from transcription to degradation. The transcription dynamics of Arc in cultured hippocampal neurons revealed two novel results: (i) A robust transcriptional burst with prolonged ON state occurs after stimulation, and (ii) transcription cycles continue even after initial stimulation is removed. The correlation of stimulation with Arc transcription and mRNA transport in individual neurons revealed that stimulus-induced Ca activity was necessary but not sufficient for triggering Arc transcription and that blocking neuronal activity did not affect the dendritic transport of newly synthesized Arc mRNAs. This mouse will provide an important reagent to investigate how individual neurons transduce activity into spatiotemporal regulation of gene expression at the synapse.
Seven neuropeptides are expressed within the Drosophila brain circadian network. Our previous mRNA profiling suggested that Allatostatin-C (AstC) is an eighth neuropeptide and specifically expressed in dorsal clock neurons (DN1s). Our results here show that AstC is, indeed, expressed in DN1s, where it oscillates. AstC is also expressed in two less well-characterized circadian neuronal clusters, the DN3s and lateral-posterior neurons (LPNs). Behavioral experiments indicate that clock-neuron-derived AstC is required to mediate evening locomotor activity under short (winter-like) and long (summer-like) photoperiods. The AstC-Receptor 2 (AstC-R2) is expressed in LNds, the clock neurons that drive evening locomotor activity, and AstC-R2 is required in these neurons to modulate the same short photoperiod evening phenotype. Ex vivo calcium imaging indicates that AstC directly inhibits a single LNd. The results suggest that a novel AstC/AstC-R2 signaling pathway, from dorsal circadian neurons to an LNd, regulates the evening phase in Drosophila.
Direct visualization of genomic loci in the 3D nucleus is important for understanding the spatial organization of the genome and its association with gene expression. Various DNA FISH methods have been developed in the past decades, all involving denaturing dsDNA and hybridizing fluorescent nucleic acid probes. Here we report a novel approach that uses in vitro constituted nuclease-deficient clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated caspase 9 (Cas9) complexes as probes to label sequence-specific genomic loci fluorescently without global DNA denaturation (Cas9-mediated fluorescence in situ hybridization, CASFISH). Using fluorescently labeled nuclease-deficient Cas9 (dCas9) protein assembled with various single-guide RNA (sgRNA), we demonstrated rapid and robust labeling of repetitive DNA elements in pericentromere, centromere, G-rich telomere, and coding gene loci. Assembling dCas9 with an array of sgRNAs tiling arbitrary target loci, we were able to visualize nonrepetitive genomic sequences. The dCas9/sgRNA binary complex is stable and binds its target DNA with high affinity, allowing sequential or simultaneous probing of multiple targets. CASFISH assays using differently colored dCas9/sgRNA complexes allow multicolor labeling of target loci in cells. In addition, the CASFISH assay is remarkably rapid under optimal conditions and is applicable for detection in primary tissue sections. This rapid, robust, less disruptive, and cost-effective technology adds a valuable tool for basic research and genetic diagnosis.
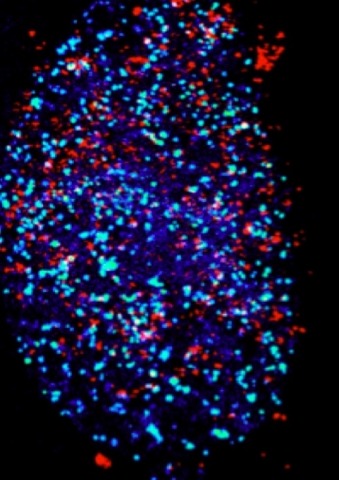
The transcriptional response of β-actin to extra-cellular stimuli is a paradigm for transcription factor complex assembly and regulation. Serum induction leads to a precisely timed pulse of β-actin transcription in the cell population. Actin protein is proposed to be involved in this response, but it is not known whether cellular actin levels affect nuclear β-actin transcription. We perturbed the levels of key signaling factors and examined the effect on the induced transcriptional pulse by following endogenous β-actin alleles in single living cells. Lowering serum response factor (SRF) protein levels leads to loss of pulse integrity, whereas reducing actin protein levels reveals positive feedback regulation, resulting in elevated gene activation and a prolonged transcriptional response. Thus, transcriptional pulse fidelity requires regulated amounts of signaling proteins, and perturbations in factor levels eliminate the physiological response, resulting in either tuning down or exaggeration of the transcriptional pulse.
Light-sheet fluorescence microscopy is able to image large specimens with high resolution by capturing the samples from multiple angles. Multiview deconvolution can substantially improve the resolution and contrast of the images, but its application has been limited owing to the large size of the data sets. Here we present a Bayesian-based derivation of multiview deconvolution that drastically improves the convergence time, and we provide a fast implementation using graphics hardware.