Filter
Associated Lab
- Aso Lab (2) Apply Aso Lab filter
- Betzig Lab (8) Apply Betzig Lab filter
- Card Lab (1) Apply Card Lab filter
- Clapham Lab (3) Apply Clapham Lab filter
- Fetter Lab (5) Apply Fetter Lab filter
- Funke Lab (6) Apply Funke Lab filter
- Harris Lab (4) Apply Harris Lab filter
- Hess Lab (80) Apply Hess Lab filter
- Jayaraman Lab (2) Apply Jayaraman Lab filter
- Lavis Lab (2) Apply Lavis Lab filter
- Lee (Albert) Lab (1) Apply Lee (Albert) Lab filter
- Lippincott-Schwartz Lab (16) Apply Lippincott-Schwartz Lab filter
- Liu (Zhe) Lab (2) Apply Liu (Zhe) Lab filter
- Looger Lab (2) Apply Looger Lab filter
- Reiser Lab (1) Apply Reiser Lab filter
- Romani Lab (1) Apply Romani Lab filter
- Rubin Lab (6) Apply Rubin Lab filter
- Saalfeld Lab (10) Apply Saalfeld Lab filter
- Scheffer Lab (11) Apply Scheffer Lab filter
- Shroff Lab (1) Apply Shroff Lab filter
- Turner Lab (1) Apply Turner Lab filter
Associated Project Team
Publication Date
- 2026 (1) Apply 2026 filter
- 2025 (7) Apply 2025 filter
- 2024 (6) Apply 2024 filter
- 2023 (5) Apply 2023 filter
- 2022 (7) Apply 2022 filter
- 2021 (5) Apply 2021 filter
- 2020 (5) Apply 2020 filter
- 2019 (5) Apply 2019 filter
- 2018 (3) Apply 2018 filter
- 2017 (4) Apply 2017 filter
- 2016 (2) Apply 2016 filter
- 2015 (7) Apply 2015 filter
- 2014 (5) Apply 2014 filter
- 2013 (2) Apply 2013 filter
- 2012 (3) Apply 2012 filter
- 2011 (2) Apply 2011 filter
- 2010 (2) Apply 2010 filter
- 2009 (2) Apply 2009 filter
- 2008 (2) Apply 2008 filter
- 2007 (1) Apply 2007 filter
- 2006 (1) Apply 2006 filter
- 1994 (2) Apply 1994 filter
- 1988 (1) Apply 1988 filter
Type of Publication
80 Publications
Showing 31-40 of 80 resultsA gas of hydrogen atoms, confined in a static magnetic trap, has been evaporatively cooled to temperatures of a few millikelvin. The initial trap configuration held the gas at 38 mK for as long as 5 h. Evaporative cooling reduced the temperature to 3.0 mK while maintaining the central density at 7.6×10 12 cm −3 . These values were determined by measurement of the rate of electronic spin relaxation and are in agreement with model calculations. Further cooling to 1 mK (inferred from the model) has been achieved. Measurements were made of the efficiency of the evaporative cooling process.
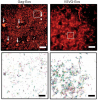
Fluorescent proteins facilitate a variety of imaging paradigms in live and fixed samples. However, they lose their fluorescence after heavy fixation, hindering applications such as correlative light and electron microscopy (CLEM). Here we report engineered variants of the photoconvertible Eos fluorescent protein that fluoresce and photoconvert normally in heavily fixed (0.5-1% OsO4), plastic resin-embedded samples, enabling correlative super-resolution fluorescence imaging and high-quality electron microscopy.
We demonstrate gas cluster ion beam scanning electron microscopy (SEM), in which wide-area ion milling is performed on a series of thick tissue sections. This three-dimensional electron microscopy technique acquires datasets with <10 nm isotropic resolution of each section, and these can then be stitched together to span the sectioned volume. Incorporating gas cluster ion beam SEM into existing single-beam and multibeam SEM workflows should be straightforward, increasing reliability while improving z resolution by a factor of three or more.
Focused Ion Beam Scanning Electron Microscopy (FIB-SEM) generates 3D datasets optimally suited for segmentation of cell ultrastructure and automated connectome tracing but is limited to small fields of view and is therefore incompatible with the new generation of ultrafast multibeam SEMs. In contrast, section-based techniques are multibeam-compatible but are limited in z-resolution making automatic segmentation of cellular ultrastructure difficult. Here we demonstrate a novel 3D electron microscopy technique, Gas Cluster Ion Beam SEM (GCIB-SEM), in which top-down, wide-area ion milling is performed on a series of thick sections, acquiring < 10 nm isotropic datasets of each which are then stitched together to span the full sectioned volume. Based on our results, incorporating GCIB-SEM into existing single beam and multibeam SEM workflows should be straightforward and should dramatically increase reliability while simultaneously improving z-resolution by a factor of 3 or more.
View Publication PageThe challenge of recovering the topology of massive neuronal circuits can potentially be met by high throughput Electron Microscopy (EM) imagery. Segmenting a 3-dimensional stack of EM images into the individual neurons is difficult, due to the low depth-resolution in existing high-throughput EM technology, such as serial section Transmission EM (ssTEM). In this paper we propose methods for detecting the high resolution locations of membranes from low depth-resolution images. We approach this problem using both a method that learns a discriminative, over-complete dictionary and a kernel SVM. We test this approach on tomographic sections produced in simulations from high resolution Focused Ion Beam (FIB) images and on low depth-resolution images acquired with ssTEM and evaluate our results by comparing it to manual labeling of this data.
We combined photoactivated localization microscopy (PALM) with live-cell single-particle tracking to create a new method termed sptPALM. We created spatially resolved maps of single-molecule motions by imaging the membrane proteins Gag and VSVG, and obtained several orders of magnitude more trajectories per cell than traditional single-particle tracking enables. By probing distinct subsets of molecules, sptPALM can provide insight into the origins of spatial and temporal heterogeneities in membranes.
Commentary: As a stepping stone to true live cell PALM (see above), our collaborator Jennifer Lippincott-Schwartz suggested using the sparse photoactivation principle of PALM to track the nanoscale motion of thousands of individual molecules within a single living cell. Termed single particle tracking PALM (sptPALM), Jennifer’s postdocs Suliana Manley and Jen Gillette used the method in our PALM rig to create spatially resolved maps of diffusion rates in the plasma membrane of live cells. sptPALM is a powerful tool to study the active cytoskeletal or passive diffusional transport of individual molecules with far more measurements per cell than is possible without sparse photoactivation.
Electron Microscopy (EM) is widely used in many scientific fields, particularly in life sciences, offering high-resolution information on the ultrastructure of biological organisms. Accurate characterization of EM image quality is important for assessing the EM tool performance, in addition to sample preparation protocol, imaging conditions, etc.This paper provides an overview of tools we developed as plugins for the popular image processing package Fiji (ImageJ) (1). These tools include signal-to-noise ratio analysis, contrast evaluation, and resolution analysis, as well as the capability to import images acquired on custom FIB-SEM instruments (2). We have also made these tools available in Python, with both versions available on GitHub.
The automated tape-collecting ultramicrotome (ATUM) makes it possible to collect large numbers of ultrathin sections quickly-the equivalent of a petabyte of high resolution images each day. However, even high throughput image acquisition strategies generate images far more slowly (at present ~1 terabyte per day). We therefore developed WaferMapper, a software package that takes a multi-resolution approach to mapping and imaging select regions within a library of ultrathin sections. This automated method selects and directs imaging of corresponding regions within each section of an ultrathin section library (UTSL) that may contain many thousands of sections. Using WaferMapper, it is possible to map thousands of tissue sections at low resolution and target multiple points of interest for high resolution imaging based on anatomical landmarks. The program can also be used to expand previously imaged regions, acquire data under different imaging conditions, or re-image after additional tissue treatments.
Many biomolecules in cells can be visualized with high sensitivity and specificity by fluorescence microscopy. However, the resolution of conventional light microscopy is limited by diffraction to ~200-250nm laterally and >500nm axially. Here, we describe superresolution methods based on single-molecule localization analysis of photoswitchable fluorophores (PALM: photoactivated localization microscopy) as well as our recent three-dimensional (3D) method (iPALM: interferometric PALM) that allows imaging with a resolution better than 20nm in all three dimensions. Considerations for their implementations, applications to multicolor imaging, and a recent development that extend the imaging depth of iPALM to ~750nm are discussed. As the spatial resolution of superresolution fluorescence microscopy converges with that of electron microscopy (EM), direct imaging of the same specimen using both approaches becomes feasible. This could be particularly useful for cross validation of experiments, and thus, we also describe recent methods that were developed for correlative superresolution fluorescence and EM.