Filter
Associated Lab
- Aguilera Castrejon Lab (19) Apply Aguilera Castrejon Lab filter
- Ahrens Lab (73) Apply Ahrens Lab filter
- Aso Lab (42) Apply Aso Lab filter
- Baker Lab (38) Apply Baker Lab filter
- Betzig Lab (116) Apply Betzig Lab filter
- Beyene Lab (15) Apply Beyene Lab filter
- Bock Lab (17) Apply Bock Lab filter
- Branson Lab (55) Apply Branson Lab filter
- Card Lab (43) Apply Card Lab filter
- Cardona Lab (64) Apply Cardona Lab filter
- Chklovskii Lab (13) Apply Chklovskii Lab filter
- Clapham Lab (16) Apply Clapham Lab filter
- Cui Lab (19) Apply Cui Lab filter
- Darshan Lab (12) Apply Darshan Lab filter
- Dennis Lab (3) Apply Dennis Lab filter
- Dickson Lab (46) Apply Dickson Lab filter
- Druckmann Lab (25) Apply Druckmann Lab filter
- Dudman Lab (56) Apply Dudman Lab filter
- Eddy/Rivas Lab (30) Apply Eddy/Rivas Lab filter
- Egnor Lab (11) Apply Egnor Lab filter
- Espinosa Medina Lab (23) Apply Espinosa Medina Lab filter
- Feliciano Lab (12) Apply Feliciano Lab filter
- Fetter Lab (41) Apply Fetter Lab filter
- FIB-SEM Technology (1) Apply FIB-SEM Technology filter
- Fitzgerald Lab (30) Apply Fitzgerald Lab filter
- Freeman Lab (15) Apply Freeman Lab filter
- Funke Lab (46) Apply Funke Lab filter
- Gonen Lab (91) Apply Gonen Lab filter
- Grigorieff Lab (62) Apply Grigorieff Lab filter
- Harris Lab (65) Apply Harris Lab filter
- Heberlein Lab (94) Apply Heberlein Lab filter
- Hermundstad Lab (30) Apply Hermundstad Lab filter
- Hess Lab (80) Apply Hess Lab filter
- Ilanges Lab (4) Apply Ilanges Lab filter
- Jayaraman Lab (48) Apply Jayaraman Lab filter
- Ji Lab (33) Apply Ji Lab filter
- Johnson Lab (6) Apply Johnson Lab filter
- Kainmueller Lab (19) Apply Kainmueller Lab filter
- Karpova Lab (14) Apply Karpova Lab filter
- Keleman Lab (13) Apply Keleman Lab filter
- Keller Lab (76) Apply Keller Lab filter
- Koay Lab (19) Apply Koay Lab filter
- Lavis Lab (158) Apply Lavis Lab filter
- Lee (Albert) Lab (34) Apply Lee (Albert) Lab filter
- Leonardo Lab (23) Apply Leonardo Lab filter
- Li Lab (32) Apply Li Lab filter
- Lippincott-Schwartz Lab (180) Apply Lippincott-Schwartz Lab filter
- Liu (Yin) Lab (8) Apply Liu (Yin) Lab filter
- Liu (Zhe) Lab (65) Apply Liu (Zhe) Lab filter
- Looger Lab (138) Apply Looger Lab filter
- Magee Lab (49) Apply Magee Lab filter
- Menon Lab (18) Apply Menon Lab filter
- Murphy Lab (13) Apply Murphy Lab filter
- O'Shea Lab (8) Apply O'Shea Lab filter
- Otopalik Lab (13) Apply Otopalik Lab filter
- Pachitariu Lab (52) Apply Pachitariu Lab filter
- Pastalkova Lab (19) Apply Pastalkova Lab filter
- Pavlopoulos Lab (19) Apply Pavlopoulos Lab filter
- Pedram Lab (15) Apply Pedram Lab filter
- Podgorski Lab (16) Apply Podgorski Lab filter
- Reiser Lab (55) Apply Reiser Lab filter
- Riddiford Lab (44) Apply Riddiford Lab filter
- Romani Lab (51) Apply Romani Lab filter
- Rubin Lab (149) Apply Rubin Lab filter
- Saalfeld Lab (64) Apply Saalfeld Lab filter
- Satou Lab (18) Apply Satou Lab filter
- Scheffer Lab (38) Apply Scheffer Lab filter
- Schreiter Lab (70) Apply Schreiter Lab filter
- Schulze Lab (1) Apply Schulze Lab filter
- Sgro Lab (23) Apply Sgro Lab filter
- Shroff Lab (31) Apply Shroff Lab filter
- Simpson Lab (23) Apply Simpson Lab filter
- Singer Lab (80) Apply Singer Lab filter
- Spruston Lab (98) Apply Spruston Lab filter
- Stern Lab (160) Apply Stern Lab filter
- Sternson Lab (54) Apply Sternson Lab filter
- Stringer Lab (41) Apply Stringer Lab filter
- Svoboda Lab (136) Apply Svoboda Lab filter
- Tebo Lab (35) Apply Tebo Lab filter
- Tervo Lab (9) Apply Tervo Lab filter
- Tillberg Lab (22) Apply Tillberg Lab filter
- Tjian Lab (64) Apply Tjian Lab filter
- Truman Lab (88) Apply Truman Lab filter
- Turaga Lab (53) Apply Turaga Lab filter
- Turner Lab (38) Apply Turner Lab filter
- Vale Lab (8) Apply Vale Lab filter
- Voigts Lab (4) Apply Voigts Lab filter
- Wang (Meng) Lab (29) Apply Wang (Meng) Lab filter
- Wang (Shaohe) Lab (25) Apply Wang (Shaohe) Lab filter
- Wong-Campos Lab (1) Apply Wong-Campos Lab filter
- Wu Lab (9) Apply Wu Lab filter
- Zlatic Lab (28) Apply Zlatic Lab filter
- Zuker Lab (25) Apply Zuker Lab filter
Associated Project Team
- CellMap (13) Apply CellMap filter
- COSEM (3) Apply COSEM filter
- FIB-SEM Technology (5) Apply FIB-SEM Technology filter
- Fly Descending Interneuron (12) Apply Fly Descending Interneuron filter
- Fly Functional Connectome (14) Apply Fly Functional Connectome filter
- Fly Olympiad (5) Apply Fly Olympiad filter
- FlyEM (56) Apply FlyEM filter
- FlyLight (50) Apply FlyLight filter
- GENIE (47) Apply GENIE filter
- Integrative Imaging (9) Apply Integrative Imaging filter
- Larval Olympiad (2) Apply Larval Olympiad filter
- MouseLight (18) Apply MouseLight filter
- NeuroSeq (1) Apply NeuroSeq filter
- ThalamoSeq (1) Apply ThalamoSeq filter
- Tool Translation Team (T3) (29) Apply Tool Translation Team (T3) filter
- Transcription Imaging (49) Apply Transcription Imaging filter
Publication Date
- 2026 (70) Apply 2026 filter
- 2025 (222) Apply 2025 filter
- 2024 (210) Apply 2024 filter
- 2023 (158) Apply 2023 filter
- 2022 (192) Apply 2022 filter
- 2021 (194) Apply 2021 filter
- 2020 (196) Apply 2020 filter
- 2019 (202) Apply 2019 filter
- 2018 (232) Apply 2018 filter
- 2017 (217) Apply 2017 filter
- 2016 (209) Apply 2016 filter
- 2015 (252) Apply 2015 filter
- 2014 (236) Apply 2014 filter
- 2013 (194) Apply 2013 filter
- 2012 (190) Apply 2012 filter
- 2011 (190) Apply 2011 filter
- 2010 (161) Apply 2010 filter
- 2009 (158) Apply 2009 filter
- 2008 (140) Apply 2008 filter
- 2007 (106) Apply 2007 filter
- 2006 (92) Apply 2006 filter
- 2005 (67) Apply 2005 filter
- 2004 (57) Apply 2004 filter
- 2003 (58) Apply 2003 filter
- 2002 (39) Apply 2002 filter
- 2001 (28) Apply 2001 filter
- 2000 (29) Apply 2000 filter
- 1999 (14) Apply 1999 filter
- 1998 (18) Apply 1998 filter
- 1997 (16) Apply 1997 filter
- 1996 (10) Apply 1996 filter
- 1995 (18) Apply 1995 filter
- 1994 (12) Apply 1994 filter
- 1993 (10) Apply 1993 filter
- 1992 (6) Apply 1992 filter
- 1991 (11) Apply 1991 filter
- 1990 (11) Apply 1990 filter
- 1989 (6) Apply 1989 filter
- 1988 (1) Apply 1988 filter
- 1987 (7) Apply 1987 filter
- 1986 (4) Apply 1986 filter
- 1985 (5) Apply 1985 filter
- 1984 (2) Apply 1984 filter
- 1983 (2) Apply 1983 filter
- 1982 (3) Apply 1982 filter
- 1981 (3) Apply 1981 filter
- 1980 (1) Apply 1980 filter
- 1979 (1) Apply 1979 filter
- 1976 (2) Apply 1976 filter
- 1973 (1) Apply 1973 filter
- 1970 (1) Apply 1970 filter
- 1967 (1) Apply 1967 filter
Type of Publication
4265 Publications
Showing 3711-3720 of 4265 resultsUnderstanding the functions of a brain region requires knowing the neural representations of its myriad inputs, local neurons and outputs. Primary visual cortex (V1) has long been thought to compute visual orientation from untuned thalamic inputs, but very few thalamic inputs have been measured in any mammal. We determined the response properties of ~28,000 thalamic boutons and ~4,000 cortical neurons in layers 1–5 of awake mouse V1. Using adaptive optics that allows accurate measurement of bouton activity deep in cortex, we found that around half of the boutons in the main thalamorecipient L4 carried orientation-tuned information and that their orientation and direction biases were also dominant in the L4 neuron population, suggesting that these neurons may inherit their selectivity from tuned thalamic inputs. Cortical neurons in all layers exhibited sharper tuning than thalamic boutons and a greater diversity of preferred orientations. Our results provide data-rich constraints for refining mechanistic models of cortical computation.
The brain contains a relatively simple circuit for forming Pavlovian associations, yet it achieves many operations common across memory systems. Recent advances have established a clear framework for learning and revealed the following key operations: ) pattern separation, whereby dense combinatorial representations of odors are preprocessed to generate highly specific, nonoverlapping odor patterns used for learning; ) convergence, in which sensory information is funneled to a small set of output neurons that guide behavioral actions; ) plasticity, where changing the mapping of sensory input to behavioral output requires a strong reinforcement signal, which is also modulated by internal state and environmental context; and ) modularization, in which a memory consists of multiple parallel traces, which are distinct in stability and flexibility and exist in anatomically well-defined modules within the network. Cross-module interactions allow for higher-order effects where past experience influences future learning. Many of these operations have parallels with processes of memory formation and action selection in more complex brains. Expected final online publication date for the , Volume 43 is July 8, 2020. Please see http://www.annualreviews.org/page/journal/pubdates for revised estimates.
We recently defined genetic traits that distinguish sympathetic from parasympathetic neurons, both preganglionic and ganglionic (Espinosa-Medina et al., Science 354:893-897, 2016). By this set of criteria, we found that the sacral autonomic outflow is sympathetic, not parasympathetic as has been thought for more than a century. Proposing such a belated shift in perspective begs the question why the new criterion (cell types defined by their genetic make-up and dependencies) should be favored over the anatomical, physiological and pharmacological considerations of long ago that inspired the "parasympathetic" classification. After a brief reminder of the former, we expound the weaknesses of the latter and argue that the novel genetic definition helps integrating neglected anatomical and physiological observations and clearing the path for future research.
Far-field optical microscopy using focused light is an important tool in a number of scientific disciplines including chemical, (bio)physical and biomedical research, particularly with respect to the study of living cells and organisms. Unfortunately, the applicability of the optical microscope is limited, since the diffraction of light imposes limitations on the spatial resolution of the image. Consequently the details of, for example, cellular protein distributions, can be visualized only to a certain extent. Fortunately, recent years have witnessed the development of 'super-resolution' far-field optical microscopy (nanoscopy) techniques such as stimulated emission depletion (STED), ground state depletion (GSD), reversible saturated optical (fluorescence) transitions (RESOLFT), photoactivation localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), structured illumination microscopy (SIM) or saturated structured illumination microscopy (SSIM), all in one way or another addressing the problem of the limited spatial resolution of far-field optical microscopy. While SIM achieves a two-fold improvement in spatial resolution compared to conventional optical microscopy, STED, RESOLFT, PALM/STORM, or SSIM have all gone beyond, pushing the limits of optical image resolution to the nanometer scale. Consequently, all super-resolution techniques open new avenues of biomedical research. Because the field is so young, the potential capabilities of different super-resolution microscopy approaches have yet to be fully explored, and uncertainties remain when considering the best choice of methodology. Thus, even for experts, the road to the future is sometimes shrouded in mist. The super-resolution optical microscopy roadmap of Journal of Physics D: Applied Physicsaddresses this need for clarity. It provides guidance to the outstanding questions through a collection of short review articles from experts in the field, giving a thorough discussion on the concepts underlying super-resolution optical microscopy, the potential of different approaches, the importance of label optimization (such as reversible photoswitchable proteins) and applications in which these methods will have a significant impact.
Insect antennae are astonishingly versatile and have multiple sensory modalities. Audition, detection of airflow, and graviception are combined in the antennal chordotonal organs. The miniaturization of these complex multisensory organs has never been investigated. Here we present a comprehensive study of the structure and scaling of the antennal chordotonal organs of the extremely miniaturized parasitoid wasp Megaphragma viggianii based on 3D electron microscopy. Johnston's organ of M. viggianii consists of 19 amphinematic scolopidia (95 cells); the central organ consists of five scolopidia (20 cells). Plesiomorphic composition includes one accessory cell per scolopidium, but in M. viggianii this ratio is only 0.3. Scolopale rods in Johnston's organ have a unique structure. Allometric analyses demonstrate the effects of scaling on the antennal chordotonal organs in insects. Our results not only shed light on the universal principles of miniaturization of sense organs, but also provide context for future interpretation of the M. viggianii connectome.
Estrogen promotes growth of estrogen receptor-positive (ER+) breast tumors. However, epidemiological studies examining the prognostic characteristics of breast cancer in postmenopausal women receiving hormone replacement therapy reveal a significant decrease in tumor dissemination, suggesting that estrogen has potential protective effects against cancer cell invasion. Here, we show that estrogen suppresses invasion of ER+ breast cancer cells by increasing transcription of the Ena/VASP protein, EVL, which promotes the generation of suppressive cortical actin bundles that inhibit motility dynamics, and is crucial for the ER-mediated suppression of invasion in vitro and in vivo. Interestingly, despite its benefits in suppressing tumor growth, anti-estrogenic endocrine therapy decreases EVL expression and increases local invasion in patients. Our results highlight the dichotomous effects of estrogen on tumor progression and suggest that, in contrast to its established role in promoting growth of ER+ tumors, estrogen has a significant role in suppressing invasion through actin cytoskeletal remodeling.
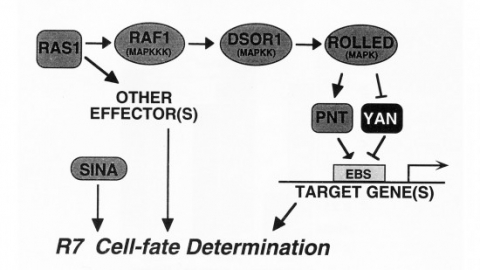
We show that the activities of two Ets-related transcription factors required for normal eye development in Drosophila, pointed and yan, are regulated by the Ras1/MAPK pathway. The pointed gene codes for two related proteins, and we show that one form is a constitutive activator of transcription, while the activity of the other form is stimulated by the Ras1/MAPK pathway. Mutation of the single consensus MAPK phosphorylation site in the second form abrogates this responsiveness. yan is a negative regulator of photoreceptor determination, and genetic data suggest that it acts as an antagonist of Ras1. We demonstrate that yan can repress transcription and that this repression activity is negatively regulated by the Ras1/MAPK signal, most likely through direct phosphorylation of yan by MAPK.
The claustrum is one of the most widely connected regions of the forebrain, yet its function has remained obscure, largely due to the experimentally challenging nature of targeting this small, thin, and elongated brain area. However, recent advances in molecular techniques have enabled the anatomy and physiology of the claustrum to be studied with the spatiotemporal and cell type-specific precision required to eventually converge on what this area does. Here we review early anatomical and electrophysiological results from cats and primates, as well as recent work in the rodent, identifying the connectivity, cell types, and physiological circuit mechanisms underlying the communication between the claustrum and the cortex. The emerging picture is one in which the rodent claustrum is closely tied to frontal/limbic regions and plays a role in processes, such as attention, that are associated with these areas. Expected final online publication date for the , Volume 43 is July 8, 2020. Please see http://www.annualreviews.org/page/journal/pubdates for revised estimates.
The ability to adjust one's behavioral strategy in complex environments is at the core of cognition. Doing so efficiently requires monitoring the reliability of the ongoing strategy and, when appropriate, switching away from it to evaluate alternatives. Studies in humans and non-human primates have uncovered signals in the anterior cingulate cortex (ACC) that reflect the pressure to switch away from the ongoing strategy, whereas other ACC signals relate to the pursuit of alternatives. However, whether these signals underlie computations that actually underpin strategy switching or merely reflect tracking of related variables remains unclear. Here we provide causal evidence that the rodent ACC actively arbitrates between persisting with the ongoing behavioral strategy and temporarily switching away to re-evaluate alternatives. Furthermore, by individually perturbing distinct output pathways, we establish that the two associated computations-determining whether to switch strategy and committing to the pursuit of a specific alternative-are segregated in the ACC microcircuitry.

The argos gene encodes a protein that is required for viability and that regulates the determination of cells in the Drosophila eye. A developmental analysis of argos mutant eyes indicates that the mystery cells, which are usually nonneuronal, are transformed into extra photoreceptors, and that supernumerary cone cells and pigment cells are also recruited. Clonal analysis indicates that argos acts nonautonomously and can diffuse over the range of several cell diameters. Conceptual translation of the argos gene suggests that it encodes a secreted protein.