Filter
Associated Lab
- Aguilera Castrejon Lab (19) Apply Aguilera Castrejon Lab filter
- Ahrens Lab (75) Apply Ahrens Lab filter
- Aso Lab (42) Apply Aso Lab filter
- Baker Lab (38) Apply Baker Lab filter
- Betzig Lab (116) Apply Betzig Lab filter
- Beyene Lab (15) Apply Beyene Lab filter
- Bock Lab (17) Apply Bock Lab filter
- Branson Lab (56) Apply Branson Lab filter
- Card Lab (43) Apply Card Lab filter
- Cardona Lab (64) Apply Cardona Lab filter
- Chklovskii Lab (13) Apply Chklovskii Lab filter
- Clapham Lab (16) Apply Clapham Lab filter
- Cui Lab (19) Apply Cui Lab filter
- Darshan Lab (12) Apply Darshan Lab filter
- Dennis Lab (3) Apply Dennis Lab filter
- Dickson Lab (46) Apply Dickson Lab filter
- Druckmann Lab (25) Apply Druckmann Lab filter
- Dudman Lab (58) Apply Dudman Lab filter
- Eddy/Rivas Lab (30) Apply Eddy/Rivas Lab filter
- Egnor Lab (11) Apply Egnor Lab filter
- Espinosa Medina Lab (25) Apply Espinosa Medina Lab filter
- Feliciano Lab (16) Apply Feliciano Lab filter
- Fetter Lab (41) Apply Fetter Lab filter
- FIB-SEM Technology (1) Apply FIB-SEM Technology filter
- Fitzgerald Lab (30) Apply Fitzgerald Lab filter
- Freeman Lab (15) Apply Freeman Lab filter
- Funke Lab (46) Apply Funke Lab filter
- Gonen Lab (91) Apply Gonen Lab filter
- Grigorieff Lab (62) Apply Grigorieff Lab filter
- Harris Lab (65) Apply Harris Lab filter
- Heberlein Lab (94) Apply Heberlein Lab filter
- Hermundstad Lab (32) Apply Hermundstad Lab filter
- Hess Lab (80) Apply Hess Lab filter
- Ilanges Lab (4) Apply Ilanges Lab filter
- Jayaraman Lab (49) Apply Jayaraman Lab filter
- Ji Lab (33) Apply Ji Lab filter
- Johnson Lab (7) Apply Johnson Lab filter
- Kainmueller Lab (19) Apply Kainmueller Lab filter
- Karpova Lab (15) Apply Karpova Lab filter
- Keleman Lab (13) Apply Keleman Lab filter
- Keller Lab (77) Apply Keller Lab filter
- Koay Lab (20) Apply Koay Lab filter
- Lavis Lab (162) Apply Lavis Lab filter
- Lee (Albert) Lab (34) Apply Lee (Albert) Lab filter
- Leonardo Lab (23) Apply Leonardo Lab filter
- Li Lab (32) Apply Li Lab filter
- Lippincott-Schwartz Lab (182) Apply Lippincott-Schwartz Lab filter
- Liu (Yin) Lab (9) Apply Liu (Yin) Lab filter
- Liu (Zhe) Lab (65) Apply Liu (Zhe) Lab filter
- Looger Lab (138) Apply Looger Lab filter
- Magee Lab (49) Apply Magee Lab filter
- Menon Lab (18) Apply Menon Lab filter
- Murphy Lab (13) Apply Murphy Lab filter
- O'Shea Lab (8) Apply O'Shea Lab filter
- Otopalik Lab (13) Apply Otopalik Lab filter
- Pachitariu Lab (56) Apply Pachitariu Lab filter
- Pastalkova Lab (19) Apply Pastalkova Lab filter
- Pavlopoulos Lab (19) Apply Pavlopoulos Lab filter
- Pedram Lab (15) Apply Pedram Lab filter
- Podgorski Lab (16) Apply Podgorski Lab filter
- Reiser Lab (55) Apply Reiser Lab filter
- Riddiford Lab (44) Apply Riddiford Lab filter
- Romani Lab (52) Apply Romani Lab filter
- Rubin Lab (149) Apply Rubin Lab filter
- Saalfeld Lab (66) Apply Saalfeld Lab filter
- Satou Lab (18) Apply Satou Lab filter
- Scheffer Lab (38) Apply Scheffer Lab filter
- Schreiter Lab (72) Apply Schreiter Lab filter
- Schulze Lab (1) Apply Schulze Lab filter
- Sgro Lab (23) Apply Sgro Lab filter
- Shroff Lab (35) Apply Shroff Lab filter
- Simpson Lab (23) Apply Simpson Lab filter
- Singer Lab (80) Apply Singer Lab filter
- Spruston Lab (98) Apply Spruston Lab filter
- Stern Lab (160) Apply Stern Lab filter
- Sternson Lab (54) Apply Sternson Lab filter
- Stringer Lab (44) Apply Stringer Lab filter
- Svoboda Lab (136) Apply Svoboda Lab filter
- Tebo Lab (36) Apply Tebo Lab filter
- Tervo Lab (10) Apply Tervo Lab filter
- Tillberg Lab (22) Apply Tillberg Lab filter
- Tjian Lab (64) Apply Tjian Lab filter
- Truman Lab (88) Apply Truman Lab filter
- Turaga Lab (53) Apply Turaga Lab filter
- Turner Lab (38) Apply Turner Lab filter
- Vale Lab (8) Apply Vale Lab filter
- Voigts Lab (5) Apply Voigts Lab filter
- Wang (Meng) Lab (31) Apply Wang (Meng) Lab filter
- Wang (Shaohe) Lab (25) Apply Wang (Shaohe) Lab filter
- Wong-Campos Lab (1) Apply Wong-Campos Lab filter
- Wu Lab (9) Apply Wu Lab filter
- Zlatic Lab (28) Apply Zlatic Lab filter
- Zuker Lab (25) Apply Zuker Lab filter
Associated Project Team
- CellMap (13) Apply CellMap filter
- COSEM (3) Apply COSEM filter
- FIB-SEM Technology (5) Apply FIB-SEM Technology filter
- Fly Descending Interneuron (12) Apply Fly Descending Interneuron filter
- Fly Functional Connectome (14) Apply Fly Functional Connectome filter
- Fly Olympiad (5) Apply Fly Olympiad filter
- FlyEM (56) Apply FlyEM filter
- FlyLight (50) Apply FlyLight filter
- GENIE (47) Apply GENIE filter
- Integrative Imaging (11) Apply Integrative Imaging filter
- Larval Olympiad (2) Apply Larval Olympiad filter
- MouseLight (18) Apply MouseLight filter
- NeuroSeq (1) Apply NeuroSeq filter
- ThalamoSeq (1) Apply ThalamoSeq filter
- Tool Translation Team (T3) (29) Apply Tool Translation Team (T3) filter
- Transcription Imaging (49) Apply Transcription Imaging filter
Publication Date
- 2026 (118) Apply 2026 filter
- 2025 (223) Apply 2025 filter
- 2024 (209) Apply 2024 filter
- 2023 (158) Apply 2023 filter
- 2022 (192) Apply 2022 filter
- 2021 (194) Apply 2021 filter
- 2020 (196) Apply 2020 filter
- 2019 (202) Apply 2019 filter
- 2018 (232) Apply 2018 filter
- 2017 (217) Apply 2017 filter
- 2016 (209) Apply 2016 filter
- 2015 (252) Apply 2015 filter
- 2014 (236) Apply 2014 filter
- 2013 (194) Apply 2013 filter
- 2012 (190) Apply 2012 filter
- 2011 (190) Apply 2011 filter
- 2010 (161) Apply 2010 filter
- 2009 (158) Apply 2009 filter
- 2008 (140) Apply 2008 filter
- 2007 (106) Apply 2007 filter
- 2006 (92) Apply 2006 filter
- 2005 (67) Apply 2005 filter
- 2004 (57) Apply 2004 filter
- 2003 (58) Apply 2003 filter
- 2002 (39) Apply 2002 filter
- 2001 (28) Apply 2001 filter
- 2000 (29) Apply 2000 filter
- 1999 (14) Apply 1999 filter
- 1998 (18) Apply 1998 filter
- 1997 (16) Apply 1997 filter
- 1996 (10) Apply 1996 filter
- 1995 (18) Apply 1995 filter
- 1994 (12) Apply 1994 filter
- 1993 (10) Apply 1993 filter
- 1992 (6) Apply 1992 filter
- 1991 (11) Apply 1991 filter
- 1990 (11) Apply 1990 filter
- 1989 (6) Apply 1989 filter
- 1988 (1) Apply 1988 filter
- 1987 (7) Apply 1987 filter
- 1986 (4) Apply 1986 filter
- 1985 (5) Apply 1985 filter
- 1984 (2) Apply 1984 filter
- 1983 (2) Apply 1983 filter
- 1982 (3) Apply 1982 filter
- 1981 (3) Apply 1981 filter
- 1980 (1) Apply 1980 filter
- 1979 (1) Apply 1979 filter
- 1976 (2) Apply 1976 filter
- 1973 (1) Apply 1973 filter
- 1970 (1) Apply 1970 filter
- 1967 (1) Apply 1967 filter
Type of Publication
4313 Publications
Showing 431-440 of 4313 resultsSmall molecule fluorophores are essential tools for chemical biology. A benefit of synthetic dyes is the ability to employ chemical approaches to control the properties and direct the position of the fluorophore. Applying modern synthetic organic chemistry strategies enables efficient tailoring of the chemical structure to obtain probes for specific biological experiments. Chemistry can also be used to activate fluorophores; new fluorogenic enzyme substrates and photoactivatable compounds with improved properties have been prepared that facilitate advanced imaging experiments with low background fluorescence. Finally, chemical reactions in live cells can be used to direct the spatial distribution of the fluorophore, allowing labeling of defined cellular regions with synthetic dyes.
Neurobiological processes occur on spatiotemporal scales spanning many orders of magnitude. Greater understanding of these processes therefore demands improvements in the tools used in their study. Here we review recent efforts to enhance the speed and resolution of one such tool, fluorescence microscopy, with an eye toward its application to neurobiological problems. On the speed front, improvements in beam scanning technology, signal generation rates, and photodamage mediation are bringing us closer to the goal of real-time functional imaging of extended neural networks. With regard to resolution, emerging methods of adaptive optics may lead to diffraction-limited imaging or much deeper imaging in optically inhomogeneous tissues, and super-resolution techniques may prove a powerful adjunct to electron microscopic methods for nanometric neural circuit reconstruction.
Neurobiological processes occur on spatiotemporal scales spanning many orders of magnitude. Greater understanding of these processes therefore demands improvements in the tools used in their study. Here we review recent efforts to enhance the speed and resolution of one such tool, fluorescence microscopy, with an eye toward its application to neurobiological problems. On the speed front, improvements in beam scanning technology, signal generation rates, and photodamage mediation are bringing us closer to the goal of real-time functional imaging of extended neural networks. With regard to resolution, emerging methods of adaptive optics may lead to diffraction-limited imaging or much deeper imaging in optically inhomogeneous tissues, and super-resolution techniques may prove a powerful adjunct to electron microscopic methods for nanometric neural circuit reconstruction.
Commentary: A brief review of recent trends in microscopy. The section “Caveats regarding the application of superresolution microscopy” was written in an effort to inject a dose of reality and caution into the unquestioning enthusiasm in the academic community for all things superresolution, covering the topics of labeling density and specificity, sample preparation artifacts, speed vs. resolution vs. photodamage, and the implications of signal-to-background for Nyquist vs. Rayleigh definitions of resolution.
Kainic acid-induced status epilepticus (KA-SE) in mature rats results in the development of spontaneous recurrent seizures and a pattern of cell death resembling hippocampal sclerosis in patients with temporal lobe epilepsy. In contrast, KA-SE in young animals before postnatal day (P) 18 is less likely to cause cell death or epilepsy. To investigate whether changes in neuronal excitability occur in the subiculum after KA-SE, we examined the age-dependent effects of SE on the bursting neurons of subiculum, the major output region of the hippocampus. Patch-clamp recordings were used to monitor bursting in pyramidal neurons in the subiculum of rat hippocampal slices. Neurons were studied either one or 2-3 weeks following injection of KA or saline (control) in immature (P15) or more mature (P30) rats, which differ in their sensitivity to KA as well as the long-term sequelae of the KA-SE. A significantly greater proportion of subicular pyramidal neurons from P15 rats were strong-bursting neurons and showed increased frequency-dependent bursting compared to P30 animals. Frequency-dependent burst firing was enhanced in P30, but not in P15 rats following KA-SE. The enhancement of bursting induced by KA-SE in more mature rats suggests that the frequency-dependent limitation of repetitive burst firing, which normally occurs in the subiculum, is compromised following SE. These changes could facilitate the initiation of spontaneous recurrent seizures or their spread from the hippocampus to other parts of the brain.
Often referred to as a 'fight,' survival involves intense competition over resources. Threat displays and high-intensity attacks are just a few of the aggressive actions exhibited during these contests. Certain motor programs are species-specific, like the vibration of a rattlesnake tail. However, conserved behavioral features are found across species, which appear to be mirrored within the brain. Further parallels have been found across sexes between aggression-promoting contexts and the underlying neuronal circuits. Unraveling the complex web of conserved and variable circuit mechanisms has been considerably advanced by the generation of brain-wiring diagrams in adult female and male Drosophila melanogaster. Here, I will summarize current research, primarily in Drosophila, on how contexts, sensory cues, and internal states regulate aggression across sexes.
Organismal aging involves functional declines in both somatic and reproductive tissues. Multiple strategies have been discovered to extend lifespan across species. However, how age-related molecular changes differ among various tissues and how those lifespan-extending strategies slow tissue aging in distinct manners remain unclear. Here we generated the transcriptomic Cell Atlas of Worm Aging (CAWA, http://mengwanglab.org/atlas ) of wild-type and long-lived strains. We discovered cell-specific, age-related molecular and functional signatures across all somatic and germ cell types. We developed transcriptomic aging clocks for different tissues and quantitatively determined how three different pro-longevity strategies slow tissue aging distinctively. Furthermore, through genome-wide profiling of alternative polyadenylation (APA) events in different tissues, we discovered cell-type-specific APA changes during aging and revealed how these changes are differentially affected by the pro-longevity strategies. Together, this study offers fundamental molecular insights into both somatic and reproductive aging and provides a valuable resource for in-depth understanding of the diversity of pro-longevity mechanisms.
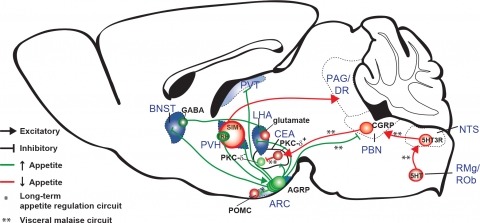
New tools for mapping and manipulating molecularly defined neural circuits have improved understanding of how the central nervous system regulates appetite. Studies focused on AGRP neurons, a starvation-sensitive hypothalamic population, have identified multiple circuit elements that can elicit or suppress feeding behavior. Distinct axon projections of this neuron population point to different circuits that regulate long-term appetite, short-term feeding, or visceral malaise-mediated anorexia. Here, we review recent studies examining these neural circuits that control food intake. © 2014 S. Karger AG, Basel.
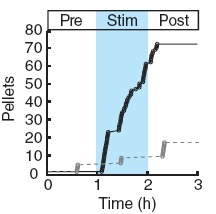
Two intermingled hypothalamic neuron populations specified by expression of agouti-related peptide (AGRP) or pro-opiomelanocortin (POMC) positively and negatively influence feeding behavior, respectively, possibly by reciprocally regulating downstream melanocortin receptors. However, the sufficiency of these neurons to control behavior and the relationship of their activity to the magnitude and dynamics of feeding are unknown. To measure this, we used channelrhodopsin-2 for cell type-specific photostimulation. Activation of only 800 AGRP neurons in mice evoked voracious feeding within minutes. The behavioral response increased with photoexcitable neuron number, photostimulation frequency and stimulus duration. Conversely, POMC neuron stimulation reduced food intake and body weight, which required melanocortin receptor signaling. However, AGRP neuron-mediated feeding was not dependent on suppressing this melanocortin pathway, indicating that AGRP neurons directly engage feeding circuits. Furthermore, feeding was evoked selectively over drinking without training or prior photostimulus exposure, which suggests that AGRP neurons serve a dedicated role coordinating this complex behavior.
A decline in ocular lens transparency known as cataract afflicts 90% of individuals by the age 70. Chronic deterioration of lens tissue occurs as a pathophysiological consequence of defective water and nutrient circulation through channel and transporter proteins. A key component is the aquaporin-0 (AQP0) water channel whose permeability is tightly regulated in healthy lenses. Using a variety of cellular and biochemical approaches we have discovered that products of the A-kinase anchoring protein 2 gene (AKAP2/AKAP-KL) form a stable complex with AQP0 to sequester protein kinase A (PKA) with the channel. This permits PKA phosphorylation of serine 235 within a calmodulin (CaM)-binding domain of AQP0. The additional negative charge introduced by phosphoserine 235 perturbs electrostatic interactions between AQP0 and CaM to favour water influx through the channel. In isolated mouse lenses, displacement of PKA from the AKAP2-AQP0 channel complex promotes cortical cataracts as characterized by severe opacities and cellular damage. Thus, anchored PKA modulation of AQP0 is a homeostatic mechanism that must be physically intact to preserve lens transparency.